INTRODUÇÃO
As dermatoses neutrofílicas agudas, grupo de doenças que inclui o Síndrome de Sweet (SS) e o Pioderma Gangrenosum (PG), são entidades raras cujo diagnóstico definitivo nem sempre é claro, por partilharem alguns aspetos da fisiopatologia, clínica, condições associadas, achados laboratoriais, histopatológicos e resposta favorável à corticoterapia. Autores defendem que constituem diferentes expressões de gravidade da mesma doença.
DESCRIÇÃO DO CASO
Apresentamos o caso de uma mulher, 25 anos, obesa, sem outros antecedentes, que recorreu ao SU devido ao aparecimento súbito de pápulas e placas eritemato-purpúricas, dolorosas (sensação de queimadura) e não pruriginosas, com dimensões variando entre poucos milímetros e vários centímetros, algumas confluentes; nalgumas pápulas era visível uma pústula central. Duas das lesões (parede abdominal e coxa) evoluíram rapidamente para úlceras com cerca de 10x6 e 8x4cm, de bordo violáceo, bem definido e fundo necrótico. As lesões localizavam-se de forma assimétrica na região abdominal inferior, metade inferior do dorso, raiz dos membros inferiores e nádegas e acompanhavam-se de febre alta. Três dias antes do aparecimento da dermatose a doente referia início de odinofagia, mialgias, cefaleias e tosse seca, tendo sido medicada com ibuprofeno e amoxicilina/clavulanato. Analiticamente apresentava leucocitose (~25.000/uL) neutrofílica e elevação da PCR (~200mg/L). Foi internada e iniciou prednisolona ~1mg/kg/dia (80mg/d) e, pela presunção de sobreinfecção, ceftriaxone e clindamicina. A evolução foi rapidamente favorável, com apirexia e normalização dos marcadores de inflamação. A biópsia cutânea revelou infiltrado inflamatório neutrofílico intenso, com participação de eosinófilos e formação de pústulas intra-epidérmicas e sub-epidérmicas, sem evidência de fenómenos vasculíticos. Num dos fragmentos de biópsia observavam-se alterações necróticas em toda a espessura dérmica. Foi orientada para consulta de Medicina Interna e Dermatologia, tendo-se verificado cicatrização lenta das lesões, que evoluíram com formação de cicatriz. Conseguida a descontinuação de corticóide ao longo de 3 meses. A história clínica exaustiva, o exame físico e os exames complementares não identificaram doença sistémica associada, nomeadamente neoplasias hematológicas ou sólidas, doença inflamatória intestinal (DII), doenças auto-imunes ou relação com fármacos. Ainda que a associação com a infeção das vias respiratórias superiores (VRS) fosse tentadora, no ano seguinte assistiu-se a duas recidivas, de menor gravidade (pequenas papulo-pústulas do tronco e membros superiores), que não foram precedidas por quadro infecioso, pelo que assumimos que estamos perante um caso idiopático.
DISCUSSÃO
Existe um conjunto de características comuns, e outras distintivas, entre SS e PG. Pensa-se que reacções de hipersensibilidade (a antigénios microbianos ou tumorais) e desregulações do sistema imune (associação frequente com doenças auto-imunes como DII e artropatias inflamatórias) podem contribuir para a ativação dos neutrófilos verificada em ambas as condições, mas permanecem ainda por explicar os mecanismos fisiopatológicos precisos.
As lesões cutâneas de ambas as condições parecem ser mediadas por neutrófilos. As características histopatológicas, a leucocitose periférica e a associação com a administração de G-CSF suportam a importância dos neutrófilos e das quimiocinas endógenas na patogénese de ambas as doenças[1]. O aumento da produção de G-CSF pelas células tumorais foi também um mecanismo proposto na doença associada a neoplasias[2, 3]. O TNF foi também implicado em alguns estudos e a resposta ao infliximab e a outros anti-TNF suporta o seu papel na fisiopatologia[4].
Epidemiologia e doenças associadas
Ambas são doenças raras, com maior incidência em adultos de 30-60 anos e predileção pelo sexo feminino, sobretudo na forma clássica de SS[2]. O SS associa-se a um amplo espectro de doenças, subdividindo-se em SS clássico, SS associado a neoplasias e SS associado a fármacos. O SS clássico/idiopático é a forma mais frequente de SS, é mais comum em mulheres e pode ocorrer associado a infeções (gastrointestinais ou das VRS, 1-3 semanas após a infecção), DII e gravidez[2]. As neoplasias mais frequentemente associadas são as hematológicas, sendo a leucemia mielóide aguda a mais frequente. Entre os tumores sólidos, destacam-se os carcinomas genito-urinários, gastro-intestinais e da mama. De entre os múltiplos fármacos relacionados com o SS, o G-CSF é o que tem uma associação mais evidente[5]. O PG associa-se a doença sistémica em mais de 50% dos casos, sendo as mais frequentes DII, doenças hematológicas (mais frequentemente gamapatia monoclonal IgA) e artropatias (artrite reumatóide e artrites seronegativas)[4]. Ambos podem preceder ou seguir-se ao diagnóstico de uma destas doenças, podendo ou não acompanhar o seu curso clínico.
Clínica
Tipicamente, SS e PG têm manifestações cutâneas distintas que permitem orientar o diagnóstico diferencial, mas podem ocorrer variantes e manifestações atípicas que contribuem para maior complexidade diagnóstica. O SS idiopático/clássico caracteriza-se pelo aparecimento de pápulas, placas e nódulos eritematosos e dolorosos, não pruriginosos, com tamanho entre poucos milímetros a vários centímetros de diâmetro e superfície com aspeto designado de pseudo-vesiculação. As lesões podem-se tornar bolhosas e ulceradas (clinicamente semelhantes às lesões clássicas de PG, sendo este aspecto mais frequente quando há associação com doenças malignas). Na presença de neoplasias hematológicas pode ocorrer ulceração da mucosa oral, incomum na forma clássica do SS[2].
O PG clássico/ulcerativo (a forma clínica mais frequente) caracteriza-se pelo aparecimento de uma pápula, pústula, ou nódulo inflamatório, doloroso (por vezes com dor desproporcional ao aspeto da lesão), que rapidamente se expande, levando à formação de úlcera. A lesão pode ser única ou múltipla e pode haver lesões em diferentes fases de evolução. A úlcera pode estender-se até à gordura subcutânea e à fáscia e a sua base é geralmente purulenta e necrótica. O fenómeno de patergia (indução ou exacerbação das lesões em locais de traumatismo) pode ocorrer tanto no PG como no SS, embora esta característica não seja essencial ao diagnóstico[2]. O SS e o PG bolhosos, menos frequentes, caracterizam-se por vesículas e bolhas flácidas sobre placas eritematosas que evoluem para ulceração. Os subtipos bolhosos associam-se mais frequentemente a neoplasias hematológicas.
A distribuição das lesões pode ajudar no diagnóstico diferencial das formas clássicas o SS é geralmente assimétrico, envolvendo preferencialmente a cabeça, o pescoço e os membros superiores, enquanto o PG ulcerativo envolve mais frequentemente os membros inferiores. A distribuição das lesões é distinta no PG bolhoso e pustuloso (membros superiores e face) e no SS associado a fármacos e neoplasias (extremidades inferiores). A rapidez de evolução das lesões pode igualmente contribuir para a distinção entre as duas entidades as lesões de SS caracterizam-se pelo seu aparecimento súbito, enquanto no PG é típico o aparecimento de uma lesão (pápula, vesícula ou nódulo) que evolui em poucos dias para formação de erosão/úlcera. Além das lesões cutâneas, em ambas as situações é comum a presença de febre >38ºC e pode haver sinais e sintomas sugestivos de doença associada. Dez a 20% dos doentes com SS clássico e associado a neoplasias podem-se apresentar sem febre[2].
Achados laboratoriais e histopatológicos
As alterações laboratoriais mais frequentemente encontradas são a leucocitose neutrofílica e a elevação da PCR e da VS. Anemia e alterações plaquetárias podem ser encontradas nos casos de doença hematológica e no SS associado a fármacos. Para identificação de doenças associadas podem ser efetuados exames auxiliares adicionais, guiados pela clínica e pelo contexto individual.
A biópsia cutânea é fundamental sempre que exista dúvida quanto ao diagnóstico. No SS os achados típicos incluem edema da derme superficial e infiltrado neutrofílico denso na derme superior e média (ocasionalmente com participação de eosinófilos), poupando a epiderme; pode haver edema endotelial, mas tipicamente não existem fenómenos de vasculite (embora a sua presença não exclua o diagnóstico)[6]. No PG a biópsia cutânea demonstra geralmente necrose da epiderme e derme superficial juntamente com infiltrado inflamatório misto, podendo ocasionalmente observar-se fenómenos de vasculite.
Na tabela 1 apresentamos os critérios de diagnóstico aceites pela maioria dos autores[7-9]. Em ambos se considera que são necessários 2 critérios major e 2 minor para estabelecer o diagnóstico.
Tratamento e prognóstico
Em ambas as dermatoses a terapêutica tem como objectivo suprimir o processo inflamatório e proporcionar ambiente favorável à cicatrização através de cuidados locais. Intervenções locais podem ser suficientes em casos de doença leve/moderada, estando a corticoterapia sistémica (prednisolona ~1mg/kg/d) indicada na doença mais extensa. No SS os sintomas geralmente melhoram em 48h e as lesões cutâneas resolvem sem cicatriz em 1-3 semanas, a menos que tenha existido ulceração[7,10]. No PG podem-se observar sinais de melhoria dias após início do tratamento, mas a cicatrização completa da úlcera geralmente demora semanas a meses e cursa com cicatriz residual[11,12].A redução da corticoterapia deve ser guiada pela evolução clínica, iniciando-se geralmente às 4 semanas[11]. Podem ocorrer recidivas, mesmo após anos de doença quiescente. Salienta-se que a resposta favorável à corticoterapia integra os critérios de diagnóstico de ambas as dermatoses.
COMENTÁRIO FINAL
O caso relatado, com aspetos sugestivos de SS (o aspecto das lesões de menores dimensões, o facto de serem múltiplas) e outros de PG (a dimensão das lesões ulceradas, a sua localização, a lentidão da resposta ao tratamento e a necrose dérmica extensa), evidencia a dificuldade de diagnóstico diferencial entre as duas condições, mesmo na posse de elementos clínicos, laboratoriais e histopatológicos. Os critérios de diagnóstico de ambas as situações não permitem afirmar ou excluir SS/PG, principalmente em casos com manifestações atípicas. A existência de mecanismos fisiopatológicos comuns, embora ainda incompletamente esclarecidos, levanta a hipótese de que estas condições podem constituir diferentes espectros de gravidade da mesma doença.
Quadro I
Critérios de diagnóstico de SS e PG
| | | |
| | Síndrome de Sweet | Pioderma Gangrenosum |
| Major | Placas ou nódulos eritematosos e dolorosos de aparecimento súbito | Úlcera cutânea necrótica e dolorosa, de bordo violáceo, irregular e escavado, com progressão rápida |
| Major | Infiltrado neutrofílico denso sem evidência de vasculite | Exclusão de outras causas de ulceração cutânea |
| Minor | Febre >38ºC | Infiltração dérmica estéril por neutrófilos ± inflamação mista ± vasculite linfocítica |
| Minor | Associação com neoplasia, DII ou gravidez OU precedido por infecção das vias respiratórias, gastro-intestinal ou vacinação | Associação com DII, artrite, gamapatia IgA, neoplasias |
| Minor | Resposta favorável ao tratamento com GC sistémicos ou iodeto de potássio | Resposta favorável ao tratamento com GC sistémicos |
| Minor | Achados laboratoriais (3 dos 4): VS>20mm/h, PCR elevada, >8.000 leucócitos/uL, >70% neutrófilos | História sugestiva de patergia ou cicatriz cribriforme |
DII: doença inflamatória intestinal; GC: glucocorticóides; VS: Velocidade de sedimentação eritrocitária, PCR: Proteína C reactiva
Figura I
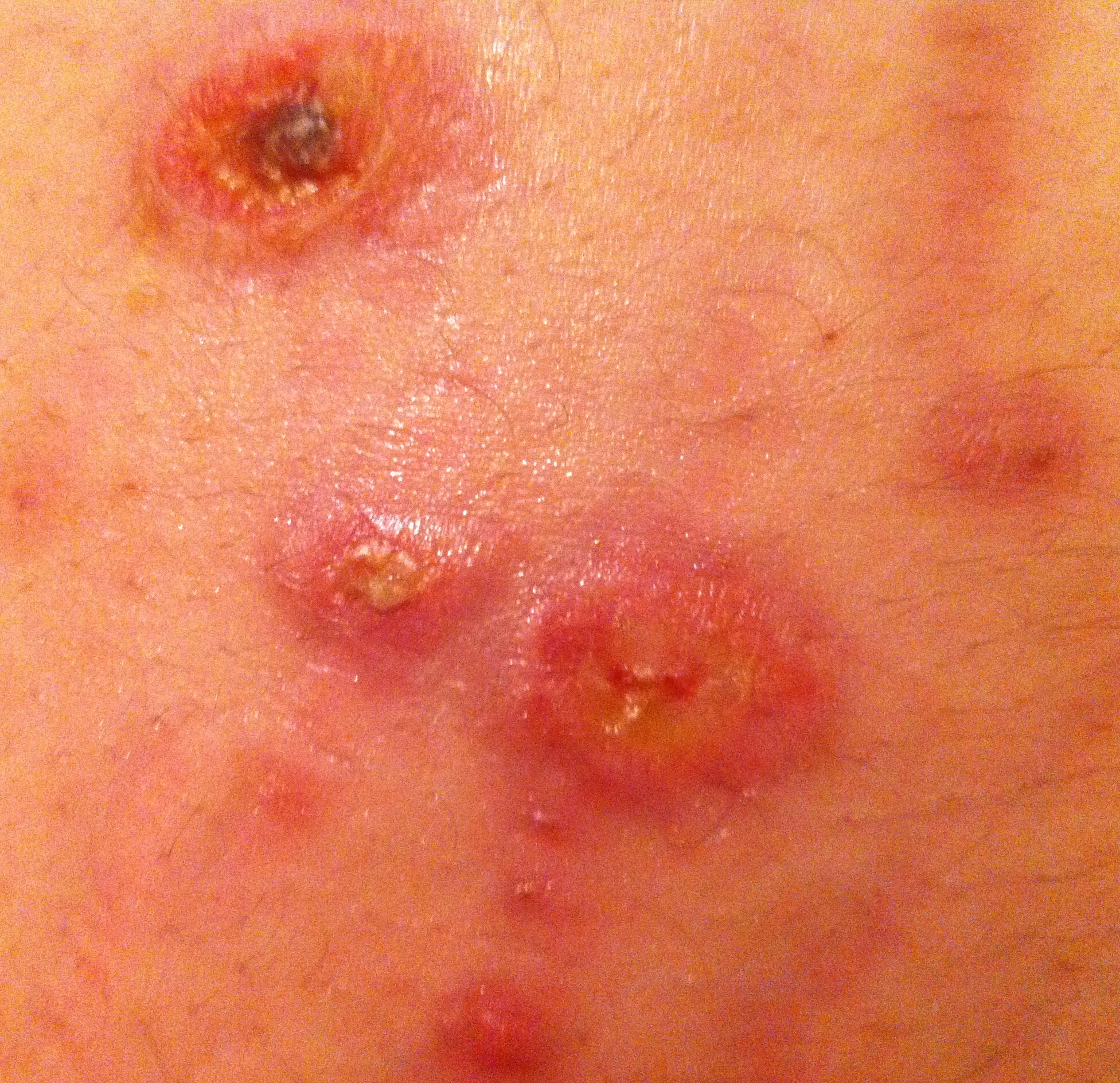
Pápulas e pequenas placas eritematosas, com vesico-pústula central (região abdominal)
Figura II
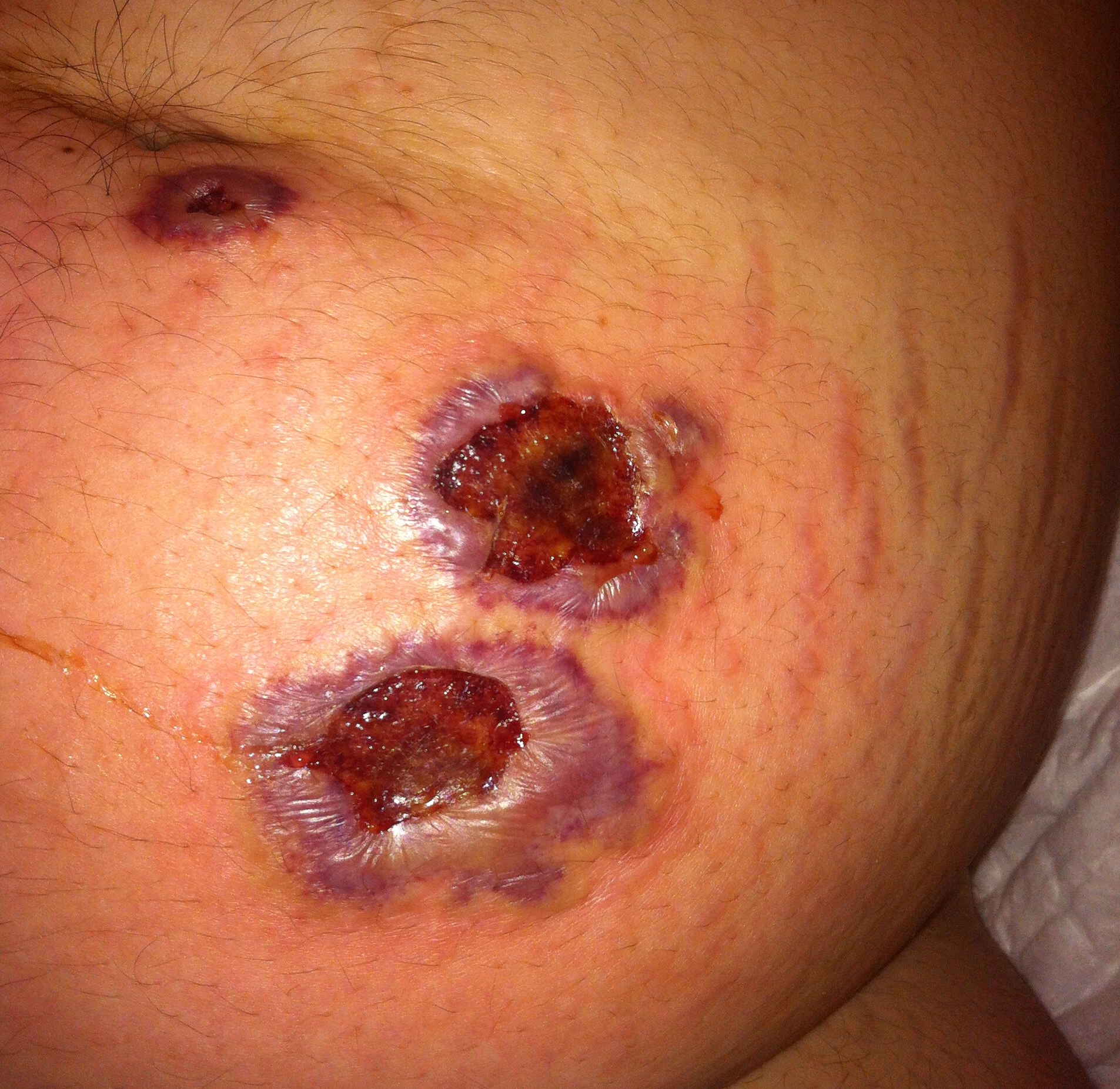
Úlceras de bordo bem definido e violáceo com fundo necrótico (região abdominal)
Figura III
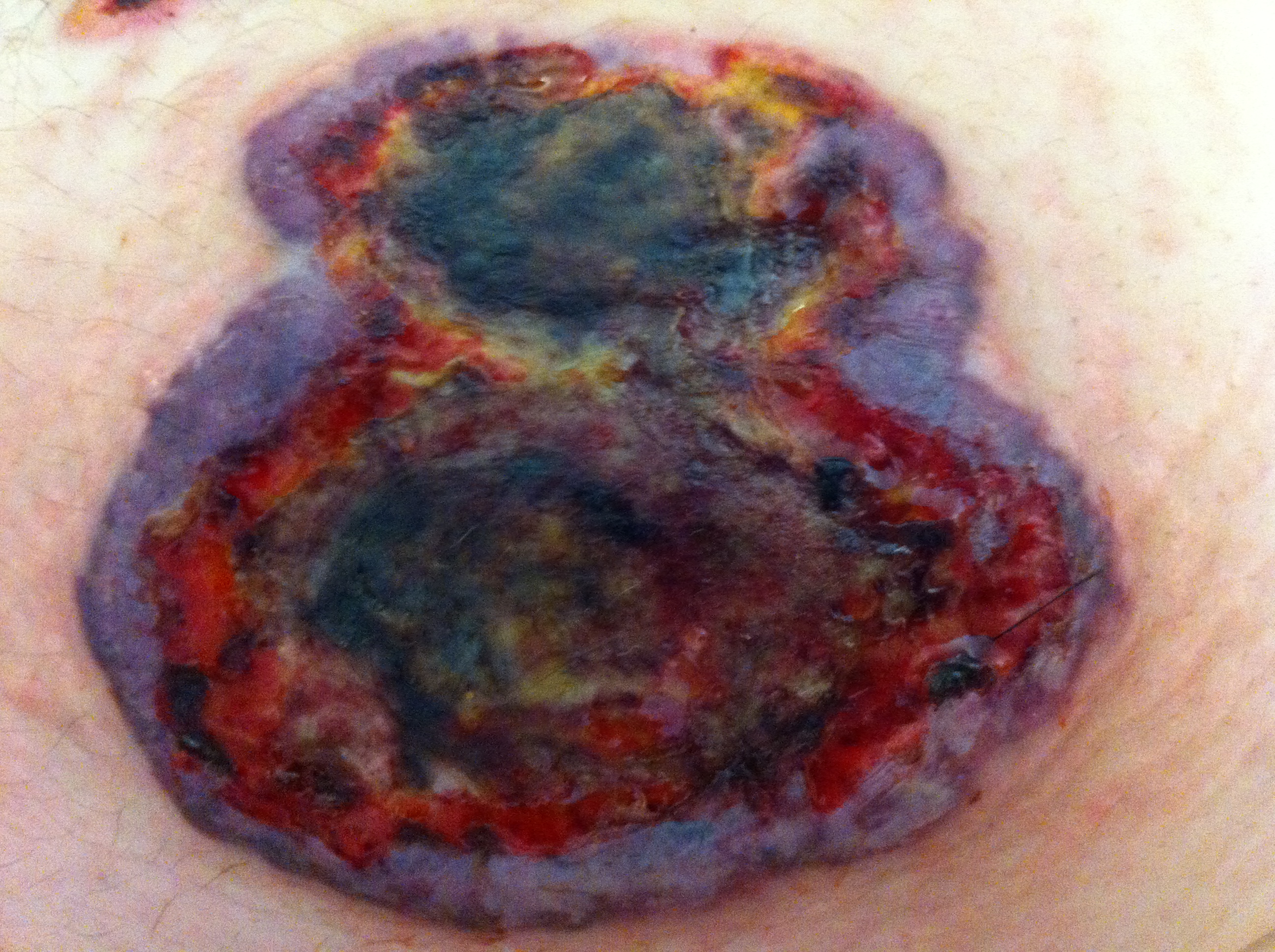
Lesão abdominal após coalescência das duas úlceras da figura 2
Figura IV
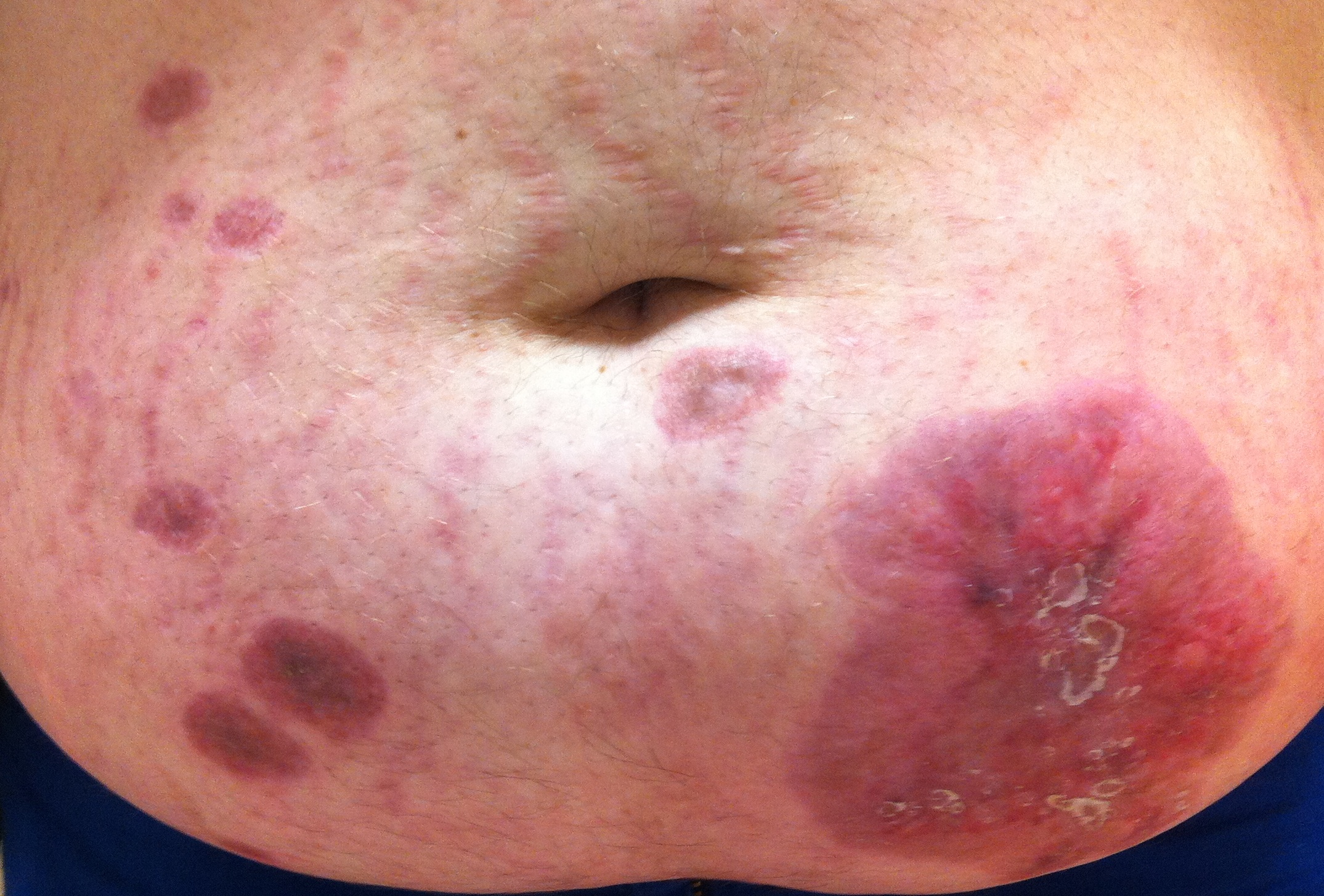
Cicatriz de úlceras, completamente reepitelizadas
Figura V
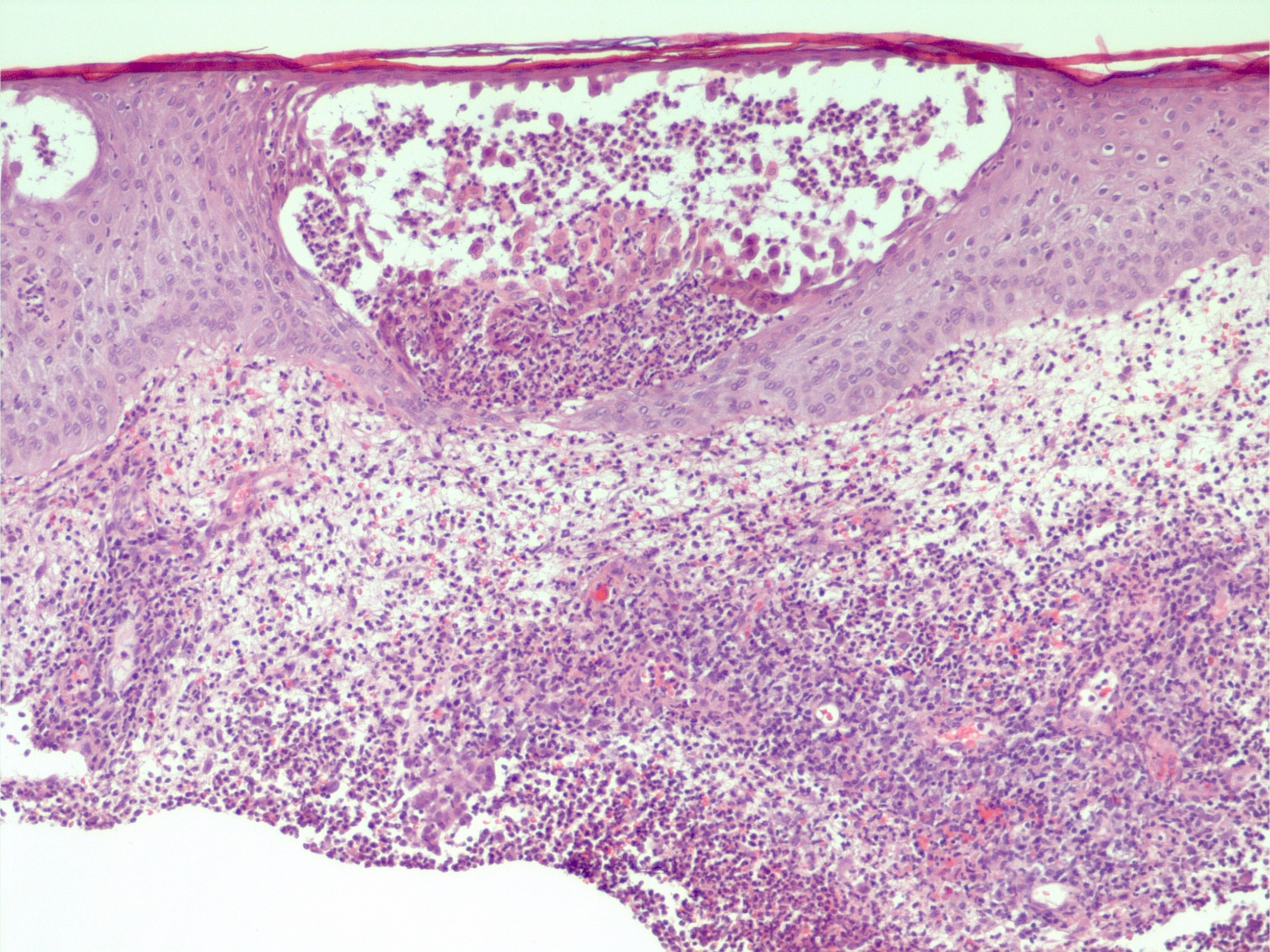
Pseudopústula intraepidérmica e infiltrado inflamatório neutrofílico
Figura VI
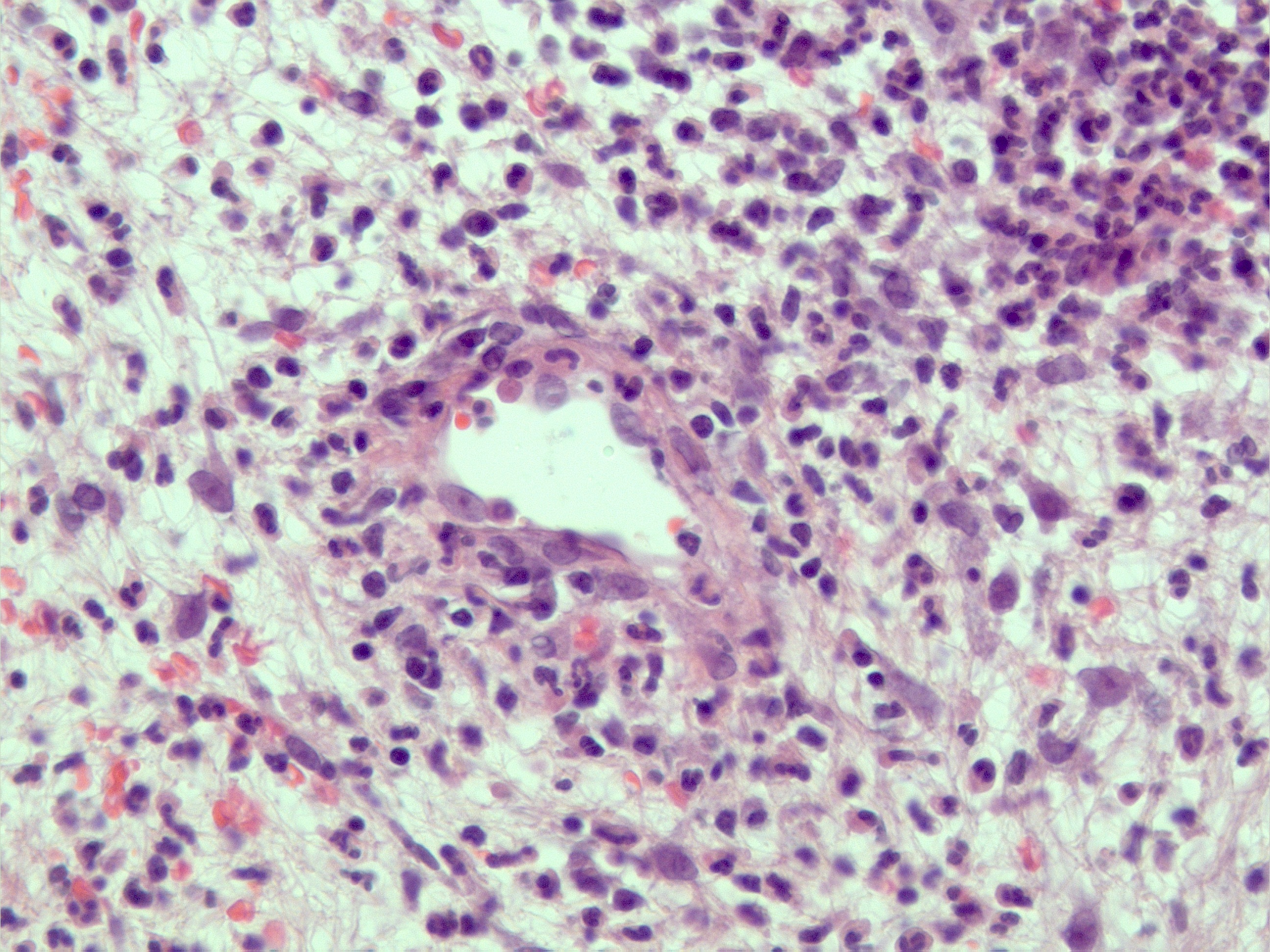
Ausência de vasculite
BIBLIOGRAFIA
1. Johnson, M.L. and R.E. Grimwood, Leukocyte colony-stimulating factors. A review of associated neutrophilic dermatoses and vasculitides. Arch Dermatol, 1994. 130(1): p. 77-81.
2. Cohen, P.R., Sweet´s syndrome--a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis, 2007. 2: p. 34.
3. Shinojima, Y., Y. Toma, and T. Terui, Sweet syndrome associated with intrahepatic cholangiocarcinoma producing granulocyte colony-stimulating factor. Br J Dermatol, 2006. 155(5): p. 1103-4.
4. Ahronowitz, I., J. Harp, and K. Shinkai, Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol, 2012. 13(3): p. 191-211.
5. White, J.M., et al., Cutaneous manifestations of granulocyte colony-stimulating factor. Clin Exp Dermatol, 2006. 31(2): p. 206-7.
6. Malone, J.C., et al., Vascular inflammation (vasculitis) in sweet syndrome: a clinicopathologic study of 28 biopsy specimens from 21 patients. Arch Dermatol, 2002. 138(3): p. 345-9.
7. von den Driesch, P., Sweet´s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol, 1994. 31(4): p. 535-56; quiz 557-60.
8. Su, W.P. and H.N. Liu, Diagnostic criteria for Sweet´s syndrome. Cutis, 1986. 37(3): p. 167-74.
9. Su, W.P., et al., Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol, 2004. 43(11): p. 790-800.
10.Cohen, P.R. and R. Kurzrock, Sweet´s syndrome: a review of current treatment options. Am J Clin Dermatol, 2002. 3(2): p. 117-31.
11. Chow, R.K. and V.C. Ho, Treatment of pyoderma gangrenosum. J Am Acad Dermatol, 1996. 34(6): p. 1047-60.
12.Ruocco, E., et al., Pyoderma gangrenosum: an updated review. J Eur Acad Dermatol Venereol, 2009. 23(9): p. 1008-17.

