A poliarterite nodosa (PAN) é uma vasculite sistémica necrotizante de vasos de médio calibre, descrita pela primeira vez por Kussmaul e Maier, em 1866.
1 Pode ser primária ou secundária, associada a infeção pelo Vírus da Hepatite B (HBV) e menos frequentemente a infeção pelos Vírus da Imunodeficiência Humana (HIV), e Hepatite C (HCV), assim como a leucemia de células pilosas.
2 A sua incidência tem vindo a diminuir, associadamente à diminuição do número de casos de infeção por HBV, variando entre 4,6 9,7 casos/milhão/ano de indivíduos da população geral. Afeta todos os grupos raciais. Tem maior predominância no sexo masculino, e maior frequência entre os 40 e 60 anos.
1,2 A etiologia da doença permanece desconhecida e ainda não existe modelo animal até à data.
1 Manifesta-se essencialmente por sintomas gerais, podendo também ter envolvimento músculo-esquelético (mialgias, fraqueza muscular, artralgias), neurológico (neuropatia periférica, lesões cerebrais), cutâneo (
livedo reticularis, nódulos, ulcerações, fenómeno de Raynaud), renal (insuficiência renal, microaneurismas das artérias renais, hipertensão arterial), cardíaco (insuficiência cardíaca, enfarte do miocárdio), gastrointestinal (hemorragias gastrointestinais, perfurações intestinais, pancreatite, enfartes ou hematomas hepáticos ou esplénicos), genital (orquite) e vascular (gangrena das extremidades, disseção da aorta ou de outras artérias de grande e médio calibre). O envolvimento pulmonar é raro e deve levantar a suspeita de infeção ou de outras vasculites.
1,2 Os critérios de diagnóstico utilizados são os da American College of Rheumatology (ACR) de 1990
3,4 (ver quadro I), tendo sido recentemente propostos os critérios da European Medicines Agency (EMEA)
5 e os do French Vasculitis Study Group (FVSG)
5, que se encontram em análise, para aplicação na prática clínica.
Caso clínico
Sexo feminino, 26 anos, raça caucasiana, com antecedentes pessoais de psoríase desde os 15 anos, abortamento espontâneo no 1º trimestre, depressão reativa e antecedente familiar de irmão com púrpura de Henoch-Schönlein na infância. Sem hábitos etílicos ou toxicómanos. Hábitos tabágicos: 9UMA.
Medicação habitual: amissulprida 50mg/dia, fluoxetina 20mg/dia e anticoncecional oral (ACO).
Assintomática até Julho de 2008, altura em que iniciou
livedo reticularis nos membros inferiores. Em Abril de 2009 teve episódio de amigdalite pultácea medicada com penicilina e desde então com episódios de febre (38ºC) de predomínio vespertino, que regrediam ao final de 4-5 dias sem terapêutica específica, associados a úlceras orais e genitais recorrentes com duração de 10 dias, astenia, anorexia e perda ponderal (8kg em 5 meses 14% do peso corporal). Em Junho de 2009, por manutenção dos sintomas realizou-se estudo analítico e foi introduzida corticoterapia (CT) 20mg/dia de prednisolona com melhoria da astenia, das úlceras orais e vaginais. Foi suspenso o ACO.
Em Agosto de 2009, com o desmame da CT para 10 mg/dia reiniciou a astenia, associada a episódios de artrite das articulações tibiotársicas e joelho, punho e cotovelo esquerdos. Foi aumentada dose de CT para 15mg/dia com melhoria clínica.
Em Setembro de 2009 iniciou tonturas e alterações do equilíbrio associadas a dificuldade de concentração, tendo sido realizada ressonância magnética nuclear crânio-encefálica (RMN-CE), onde não se observaram alterações. Foram ainda realizados o teste de Patergia e a pesquisa do HLA-B51, que se revelaram negativos. Foi observada por Oftalmologista, não apresentando lesões intraoculares. Foi aumentada dose de prednisolona para 20mg/dia, com melhoria clínica.
Posteriormente reiniciou úlceras orais recorrentes,
livedo reticularis e artrite no joelho esquerdo, não acompanhadas de úlceras genitais, alopécia ou rash malar.
Em Março de 2010, surgiu hipersudorese noturna sem febre, mialgias e fraqueza muscular de predomínio proximal e fenómeno de Raynaud. Nesta altura a perda ponderal era de 12Kg em 11 meses (21% do peso corporal). Foi admitida no Serviço de Medicina Interna para estudo, mantendo prednisolona. À admissão, a salientar palidez cutâneo-mucosa, presença de 2 placas de psoríase (1x1cm) no abdómen e
livedo reticularis nos membros inferiores (figuras nº I e II).
Do estudo etiológico realizado destacam-se: imunização para o VHB; exame direto Ziehl-Neelsen do suco gástrico negativo; serologias da sífilis, borreliose, rickettsioses e brucelose negativas; estudo da autoimunidade negativo (anticorpos anti SSA, SSB, RNP, Sm, Scl-70, JO-1, músculo liso, ANCA, antifosfolipídeos, anti-centrómero, ANA, anti DNAds e anti GBM); frações C3 e C4 e imunoglobulinas sem alterações; eritrocitúria no sedimento urinário (35 eritrócitos/campo) mas sem microalbuminúria na urina de 24h; TFG estimada de 77mL/min (fórmula de Cockcroft-Gault); função tiroideia normal; eletromiografia (EMG) dos quatro membros sem sinais de miopatia ou de polineuropatia; ecografia renovesical sem alterações relevantes; angioRMN dos vasos renais com artérias renais com padrão em rosário, nomeadamente na porção distal à esquerda (figura III). TAC-tórax de alta resolução sem evidência de áreas em vidro despolido ou cavernas.
Pela presença de perda ponderal superior a 4Kg, mialgias e fraqueza muscular,
livedo reticularis e alterações angiográficas das artérias renais firmou-se o diagnóstico de poliarterite nodosa, motivo pelo qual se iniciou ciclofosfamida EV.
Completou 6 ciclos de ciclofosfamida EV mensal. Desde então com melhoria acentuada das queixas e recuperação progressiva do peso. Após terapêutica de indução com ciclofosfamida, iniciou manutenção com azatioprina, mantendo remissão clínica sustentada. Repetiu angio-RMN dos vasos renais, com regressão completa das alterações. Em 2011, teve uma gravidez sem intercorrências, tendo sido medicada apenas com prednisolona 20mg/dia, sem recidiva. Após o parto, por apresentar hipertensão arterial e reaparecimento de fraqueza muscular, com sinais de processo inflamatório muscular na EMG, foi reintroduzida azatioprina 150mg/dia e iniciado irbesartan 150mg/dia, com melhoria clínica e bom controlo tensional.
Atualmente encontra-se grávida de 26 semanas de gestação, medicada com prednisolona 10mg/dia e clinicamente assintomática.
Discussão e conclusão
Pela clínica inicialmente apresentada poder-se-ia pensar em Doença de Behçet, pela presença de úlceras orais e genitais frequentes, e boa resposta ao corticóide. No entanto, não apresentava alterações oculares ou cutâneas típicas e os sintomas constitucionais não são frequentes na Doença de Behçet. Para infirmação desta hipótese poderia ter sido realizado o teste de Patergia, assim como o teste genético, os quais foram realizados posteriormente e cujos resultados foram negativos, o que tornou esta hipótese menos provável.
6 Outra possibilidade colocada foi o Lúpus Eritematoso Sistémico (LES), tendo em conta o quadro de sintomas constitucionais, as úlceras orais, o livedo reticularis e a artrite. Uma terceira hipótese seria uma vasculite (conjunção do
livedo reticularis, antecedente obstétrico e antecedente familiar).
4,7 Com o aparecimento das mialgias e fraqueza muscular, para além do LES e vasculites, poderiam ser pensadas miopatias inflamatórias idiopáticas e doença mista do tecido conjuntivo. Foram também equacionadas infeções (tuberculose, HIV, hepatites víricas e sífilis) e alterações metabólicas (disfunção tiroidea). Uma hipótese também a não esquecer era a miopatia induzida pelo corticóide. Foi com estas hipóteses de diagnóstico que se iniciou o estudo etiológico durante o internamento.
8 A TAC-tórax foi realizada para exclusão do envolvimento pulmonar típico das miopatias, assim como de algumas vasculites, como a poliangeíte microscópica ou o Síndrome de Churg-Strauss. A eritrocitúria poderia ser explicada em contexto de vasculite ou LES.
3,4,8 Com o estudo serológico e da autoimunidade negativos, função tiroideia e EMG normais, tornaram-se menos prováveis as miopatias inflamatórias idiopáticas, a doença mista do tecido conjuntivo, o LES, as vasculites ANCA positivas, as infeções e o hipotiroidismo. Permaneceu a hipótese de vasculites ANCA negativas, e dentro destas a PAN, que foi confirmada pela presença da alteração angiográfica renal.
3,4,7,8 Como indicado nas recomendações da European Vasculitis Study Group de 2009, realizou terapêutica de indução com corticóide e ciclofosfamida e posteriormente terapêutica de manutenção com azatioprina. Apesar da divergência de opiniões acerca da administração de ciclofosfamida por via oral (diariamente) versus intravenosa (por pulsos) optou-se pela última devido à menor incidência de efeitos adversos, nomeadamente menor toxicidade.
9 Atualmente recomenda-se que os doentes sem fatores de mau prognóstico (Five-Factor Score FFS da FVSG igual a zero) realizem terapêutica de indução com corticóide.
9 Os doentes com fatores de mau prognóstico (FFS a partir de 1: proteinúria acima de 1g/dia, insuficiência renal com creatinina acima de 1,7 mg/dl, cardiomiopatia, manifestações gastrointestinais e/ou envolvimento neurológico central) e doença ativa ou recidiva apesar do corticóide, devem realizar terapêutica de indução combinada com ciclofosfamida ou então com rituximab, que é cada vez mais preconizado (especialmente nos doentes com recidiva, doença refratária ou contraindicação à ciclofosfamida). Uma vez atingida a remissão clínica, geralmente ao final de 6-9 meses de pulsos intravenosos de ciclofosfamida ou de 3-4 meses de administração oral diária, inicia-se azatioprina ou metotrexato, sendo a duração da terapêutica de manutenção ainda incerta. Nos doentes com hepatite B deve ser utilizado um anti-retrovírico (interferon-α, lamivudina, tenofovir ou entecavir) após terapêutica com corticóide durante 2-4 semanas, seguida de plasmaferese durante 6 semanas.
2,9,10 O prognóstico depende da presença e extensão do envolvimento visceral e/ou neurológico, sendo a mortalidade maior no primeiro ano de evolução devido ao atraso no diagnóstico, não controlo da doença e complicações da terapêutica. Com terapêutica apropriada, a média de sobrevivência aos 5 anos é de 75%.
2,9,10 Este caso clínico mostra assim a complexidade do raciocínio na investigação de alguns quadros clínicos, e levanta a necessidade de pensar em patologias menos frequentes, como o caso das vasculites.
Quadro I
Critérios de diagnóstico da PAN da ACR, 1990
| Quadro I |
| Critérios de diagnóstico da PAN da ACR, 1990 |
| 1.Perda de peso superior a 4 Kg. |
| 2.Presença de livedo reticularis. |
| 3.Dor ou inflamação testicular. |
| 4.Mialgias, fraqueza ou inflamação muscular. |
| 5.Mononeuropatia ou polineuropatia. |
| 6.Hipertensão arterial diastólica superior a 90 mmHg |
| 7.Elevação da ureia (superior a 40mg/dL) ou da creatinina (superior a 1,5 mg/dL). |
| 8.Infeção pelo HBV. |
| 9.Angiografia patológica (microaneurismas). |
| 10.Biópsia das artérias de médio calibre com vasculite necrotizante, com infiltração por polimorfonucleares na fase aguda e mononucleares na fase subaguda/crónica. |
| Nota: Para estabelecer o diagnóstico de PAN é necessária a presença de pelo menos 3 critérios, sendo obrigatória a presença de um dos dois últimos. |
Figura I
Livedo reticularis.
Figura II
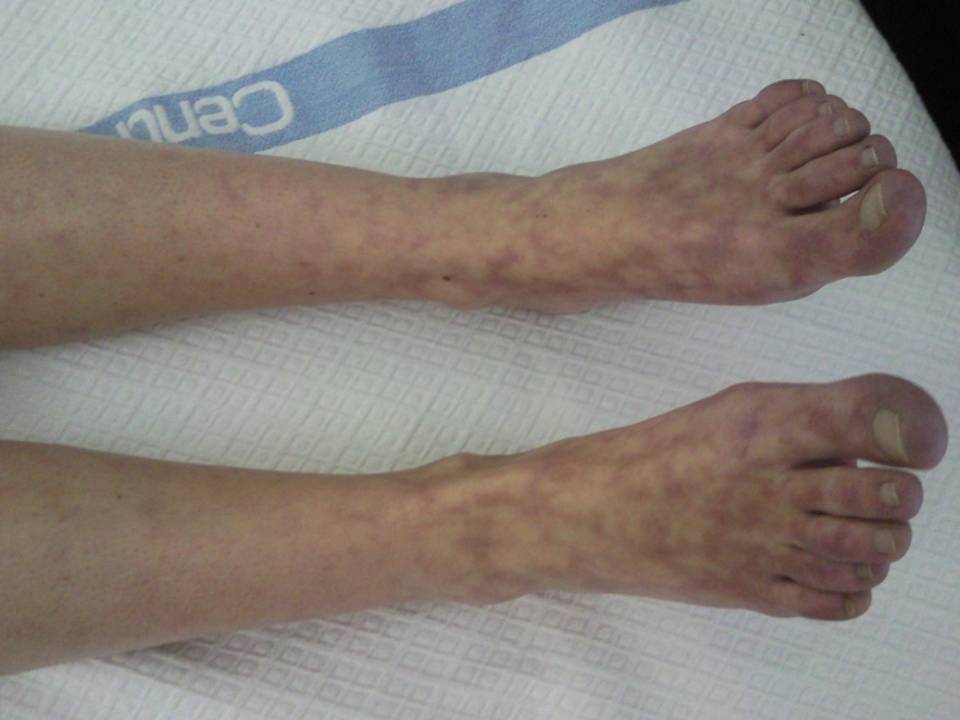
Livedo reticularis.
Figura III
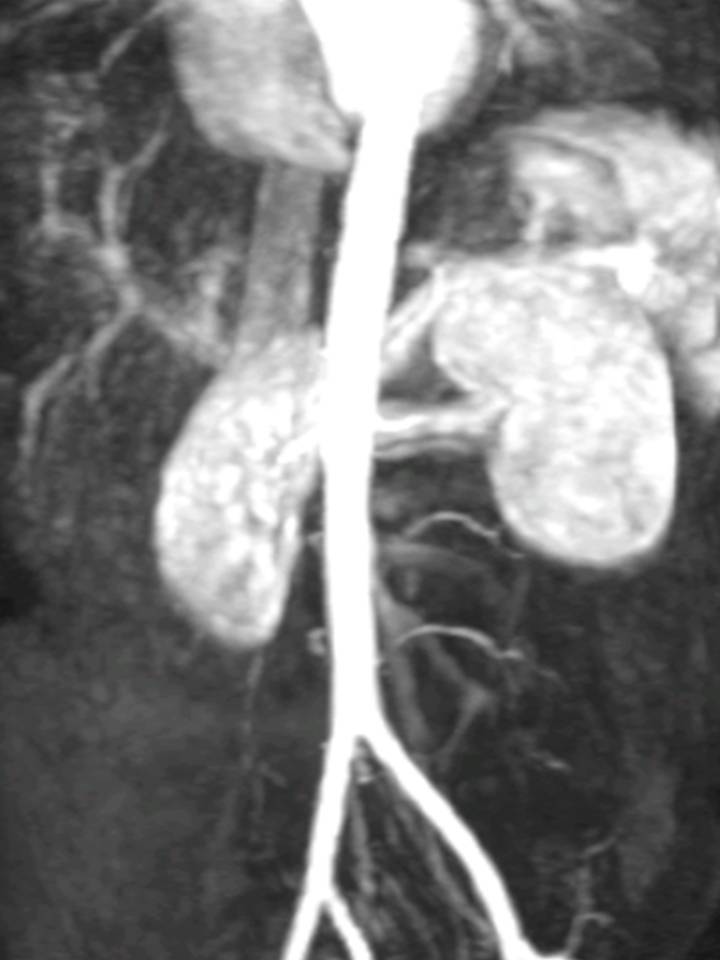
Angio-RM dos vasos renais.
BIBLIOGRAFIA
(1) Bijlsma, Pereira da Silva, Hachulla, Doherty, Cope, Lioté et al. ANCA-associated vasculitides and polyarteritis nodosa. EULAR Textbook on Rheumatic Diseases. 1st ed. Italy: BMJ Group; 2012:637-664;
(2) Kasper et al. The vasculitis syndromes. Harrison´s Internal Medicine. 18th ed. McGraw Hill; 2011;
(3) Alguire et al. Systemic Vasculitis. Medical Knowlwdge Self-assessmente Program®. 16th ed. Virginia: American College of Physicians; 2012: 64-69;
(4) Bijlsma, Pereira da Silva, Hachulla, Doherty, Cope, Lioté et al.Vasculitis: classification, secondary forms and mimics. EULAR Textbook on Rheumatic Diseases. 1st ed. Italy: BMJ Group; 2012:616-636;
(5) Abdulkader, Lane, Scott, Watts. Classification of vasculitis: EMA classification using CHCC 2012 definitions. Ann Rheum Dis 2013: 203511;
(6) Kasper et al. Kasper et al. Behçet´s Syndrome. In: Harrison´s Internal Medicine. 18th ed. McGraw Hill; 2011. Chapter 327;
(7) Kasper et al. Systemic Lupus Erythematosus. In: Harrison´s Internal Medicine. 18th ed. McGraw Hill; 2011. Chapter 319;
(8) Alguire et al. Idiopathic inflammatory myopathies. In:Medical Knowlwdge Self-assessmente Program®. 16th ed. Virginia: American College of Physicians; 2012: 58-62;
(9) European Vasculitis Study Group. EULAR Recommendations for the Management of Primary Small and Medium Vessel Vasculitis. 2009;
(10) Colburn. Vasculitides. Review of Rheumatology. Bethesda: Springer; 2012: 279-362.

