

INTRODUÇÃO
A MW foi classificada pela Organização Mundial de Saúde como uma doença linfoproliferativa de células linfoplasmocitárias que segregam imunoglobulina M (IgM), uma subcategoria dos linfomas Não-Hodgkin1,2. É uma doença rara, constituindo 2% das doenças hematológicas, com uma incidência de 3/1.000.000 indivíduos/ano3,4. Esta aumenta com a idade, apresentando uma mediana de 64 anos e é discretamente mais frequente no sexo masculino e na raça caucasiana3. A sua etiologia mantém-se desconhecida, tendo no entanto uma forte predisposição genética, sendo que familiares em primeiro grau destes doentes apresentam uma maior probabilidade, relativamente à população em geral, de desenvolver MW, linfoma não-Hodgkin, leucemia linfocítica crónica e MGUS5. Recentemente foram feitas descobertas relacionadas com a biologia e o microambiente tumoral, nomeadamente na imunomodelação6 assim como mutações genéticas ativantes em vias metabólicas determinantes na apresentação clínica e na sobrevivência global7. Aproximadamente 90% a 95% dos doentes com MW apresentam uma mutação MYD88L265P, que activa a cinase do receptor da interleucina 1 (IRAK) e a tirosina cinase de Bruton (BTK) e subsequentemente o fator de translocação nuclear kB-p65dependente, ativando vias de sinalização que contribuem para o crescimento tumoral8. Mutações nonsense no receptor de quimiocinas CXCR4 estão presentes nos casos mais agressivos de MW, inclusive na síndrome de hiperviscosidade. Estas duas mutações constituem determinantes importantes na apresentação clínica da MW e têm impacto na sobrevivência global8.
O seu início é insidioso e inespecífico, sendo o seu curso indolente. A clínica dependerá, por um lado, do grau de infiltração do tumor e por outro lado dos níveis de IgM10. Os sintomas que são mais frequentemente associados à presença de IgM monoclonal estão associados a hiperviscosidade sanguínea, a crioglobulinemia, amiloidose e a neuropatia periférica. Os sintomas típicos da infiltração tumoral são as citopénias, febre, sudorese noturna e perda ponderal, adenopatias e organomegalias10.
Englobada no conjunto das gamapatias monoclonais IgM, o diagnóstico diferencial inclui a gamapatia de significado indeterminado (MGUS), o mieloma múltiplo e outras patologias, nomeadamente doenças linfoproliferativas, amiloidose e ainda processos benignos como doenças infeciosas, inflamatórias ou auto-imunes11,12.
São aceites como critérios de diagnóstico a presença de IgM monoclonal, a infiltração da medula óssea por mais de 10% de linfócitos com diferenciação plasmocitóide e documentação por biopsia óssea de um padrão intertrabecular11.
Apesar dos avanços na terapêutica, a MW permanece incurável, apresentando uma sobrevivência mediana de 87 meses9. Os principais fatores de prognóstico associados a pior sobrevivência são idade superior a 65 anos, hemoglobina inferior ou igual a 11.5 g/dL, contagem de plaquetas inferior ou igual a 100 x 109/L, β2- microglobulina superior a 3 mg/L, concentração de proteína monoclonal no soro superior a 7.0 g/dL9. O início do tratamento é reservado para doentes com sintomas constitucionais, hiperviscosidade, neuropatia, amiloidose, crioglobulinemia sintomática, doença das aglutininas frias, assim como anemia (hemoglobina <100g/l) ou trombocitopenia (contagem plaquetas <100x109/l)13.
A estratégia terapêutica deve-se basear em características do doente e da própria doença (idade, comorbilidades, necessidade de controlo rápida da doença, se é candidato a transplante autólogo, se estão presentes citopenias, se existem complicações associadas à secreção de IgM, hiperviscosidade e presença de neuropatia). Neste contexto, a utilização de combinações de rituximab com ciclofosfamida/dexametasona, bendamustina ou ainda bortezomib/dexametasona obteve bons resultados e está indicada na maioria dos doentes14. Outras opções incluem ainda novos anticorpos monoclonais (ofatumumab), inibidores do proteossoma de segunda geração (carfilzomib), inibidores do m-TOR e inibidores da tirosina cinase de Bruton. O auto transplante de medula óssea pode ser considerado em doentes jovens com doença quimiossensível e com características de alto risco14.
CASO CLÍNICO
Apresentamos um doente do sexo masculino, de 80 anos, de raça caucasiana, com antecedentes de diverticulite complicada com perfuração (realizou sigmoidectomia em 2004) e pancitopenia (desde Janeiro 2009). Medicado em ambulatório com esomeprazol. Sem hábitos tabágicos ou alcoólicos. Sem antecedentes familiares de relevo.
Aparentemente bem até 8 de Agosto de 2009, altura em que recorreu ao SU por hematoquésia. Apresentava analiticamente uma anemia normocítica/normocrómica (hemoglobina de 5.5 gr/dl - quadro I), tendo sido internado por hemorragia digestiva alta confirmada por endoscopia digestiva alta, com necessidade de suporte transfusional. A EDA revelou ponto vermelho não sangrante mas com coágulo aderente na face anterior do antro (dieulafoy/ulcera), com aplicação de dois clips hemostáticos. O resultado anátomo-patológico da biopsia gástrica realizada revelou: Gastrite crónica atrófica quiescente com metaplasia intestinal completa (15%). H. pylori negativo. Não se observou envolvimento por doença linfoproliferativa nesta amostra.
Durante esse internamento, verificou-se um baixo rendimento transfusional, com elevação da bilirrubina indireta (0.20 mg/dl) e LDH (417 U/l), que foi interpretado como provável hemólise pós transfusional. Por episódio de hematoquésia, realizou colonoscopia total que mostrou anastomose cirúrgica aos 10 cm. Progressão até ao ileon, excluindo-se lesões sangrantes. Teve alta a 14 de Agosto, registando hemoglobina de 10,4 g/dl e bilirrubina total de 2 mg/dl (predomínio do componente indireto).
A 16 de Agosto, voltou ao SU com queixas de palidez, astenia e anorexia. Ao exame objetivo, de salientar a palidez acentuada e icterícia da pele e escleróticas. Apresentava-se hemodinamicamente estável, sem evidência de perdas hemáticas. Analiticamente, destaque para uma anemia normocítica/normocrómica, com hemoglobina 6,28 g/dl e bilirrubina total: 6,4 mg/dl (predomínio da indireta), LDH superior a 2000 U/l (quadro I). O Teste de Coombs directo foi negativo, o teste de Coombs indirecto foi positivo. Nessa altura, foi re-internado com o diagnóstico de hemólise pós transfusional, tendo efetuado nova transfusão de concentrado eritrocitário após corticoterapia.
Dos exames laboratoriais apresentou uma anemia normocrómica normocítica, leucopenia, trombocitopenia (pancitopenia), elevação da creatinina sérica, diminuição da transferrina e do ferro sérico, aumento da bilirrubina total (à custa da indireta) e aumento da beta-2-microglobulina. Na sequência destes resultados, decidiu-se avaliar exames laboratoriais anteriores, e constatou-se que análises de Janeiro e Julho do mesmo ano, efectuadas no contexto de exames de rotina pedidas pelo médico de família, apresentavam já uma pancitopenia, uma creatinina sérica progressivamente aumentada e um aumento da bilirrubina indireta (quadro I).
Laboratorialmente, a morfologia do sangue periférico não apresentava alterações. A eletroforese das proteínas revelou um pico monoclonal (figura I) IgM Kappa (quadro II). O mielograma mostrou: amostra medular compatível com infiltração medular pela MW associada a anemia das doenças crónicas. A imunofenotipagem da medula óssea revelou: 7% de células linfoides B monoclonais e 0,8% de plasmócitos monoclonais. O perfil do imunofenótipo revelou CD5+, CD10-, CD19-/+, CD20+, CD23-, CD25+, CD103- (quadro III).
A biópsia da medula óssea revelou raras células plasmáticas bifenotípicas para as cadeias kappa e lambda.
O doente realizou uma ecografia abdominal que revelou litíase vesicular múltipla, um baço de dimensões normais, mas um pouco globoso e um quisto cortical de 3,7 cm no rim esquerdo.
A presença de IgM monoclonal aumentada com aumento da cadeias livres kappa e a infiltração medular por plasmócitos, permitiu chegar ao diagnóstico definitivo de MW.
Durante o internamento iniciou o primeiro ciclo de quimioterapia com CVP (Ciclofosfamida, Vincristina e Prednisolona), sem intercorrências, apresentando apenas alguns vómitos. Dois meses depois (Novembro 2009), apresentava analiticamente hemoglobina 9.0 g/dL, leucócitos 5.1x10e3µL, neutrófilos 3.7x10e3µL linfócitos 1.1x10e3µL, plaquetas 111x10e3µL, creatinina 2.4 mg/dL e LDH 295 U/L. Considerou-se a macroglobulinemia refratária ao tratamento com CVP e em consulta de grupo de Hemato-Oncologia decidiu-se iniciar tratamento com R-CHOP (Rituximab, Ciclofosfamida, Hidroxidoxorubicina - doxorubicina, Vincristina - Oncovin, Prednisona).
Em Maio de 2010, completou oito ciclos com R-CHOP, analiticamente com hemoglobina de 11 g/dl, leucócitos de 4.5x10e3µL, neutrófilos de 2.9 x10e3µL e plaquetas de 204 x10e3µL. Como intercorrências, destaque para dois episódios de rectorragias, podendo estes dever-se à síndrome de hiperviscosidade. Complementarmente, efetuou estudo com EDA, colonoscopia e videoenteroscopia por cápsula, todas sem alterações.
Em consulta de follow-up de Hemato-Oncologia apresentou-se sem perdas hemáticas e analiticamente com cinética de ferro normal, com valores de hemoglobina sempre estáveis (10 - 10.8 g/dl - anemia normocítica/normocrómica) e insuficiência renal crónica. Como episódios cirúrgicos, destaque para uma apendicite aguda, submetido a apendicectomia e ainda uma colecistite aguda litíásica submetida a colecistectomia, sem intercorrências (Maio e Julho 2012). Em Outubro de 2013, na última consulta de Hemato-Oncologia apresentava uma hemoglobina de 12.8 g/dL, leucócitos 10.1x10e3µL e plaquetas 117x10e3µL., sem queixas de novo. O último registo informático revela um internamento em Novembro de 2013 por edema agudo do pulmão em contexto de crise hipertensiva e uma infeção respiratória nosocomial. Teve alta para o domicílio. Tivemos conhecimento do seu falecimento, cuja causa de morte se desconhece, 52 meses após o diagnóstico.
DISCUSSÃO E CONCLUSÕES
A MW é uma doença linfoproliferativa rara, que afeta doentes de idade avançada, apresentando uma incidência ligeiramente aumentada no sexo masculino e na raça caucasiana. O quadro clínico é insidioso e inespecífico. A imunofenotipagem dos linfócitos B e plasmócitos era concordante com o descrito na literatura, como suportando mas não necessários para o diagnóstico de MW15. A gamapatia monoclonal IgM, mais frequentemente associada com a MW, impõe múltiplas hipóteses diagnósticas, sendo geralmente essa a primeira dificuldade na abordagem destes doentes. Muitas vezes, a interposição de características clínicas não permite que o diagnóstico seja exclusivamente apoiado em critérios clínicos.
A dificuldade do nosso caso clínico prendeu-se, sobretudo, em enquadrar o doente como um todo e inserido num contexto clínico. Este foi mascarado pela intercorrência de hemorragia digestiva alta com anemia e necessidade de suporte transfusional, associado a uma hiperbilirrubinemia indireta, induzindo erradamente ao diagnóstico de hemólise pós-transfusional. Esta hipótese plausível de diagnóstico minimizou a suspeição de outros diagnósticos e a formulação de uma investigação clínica completa nesse contexto que conduziu ao diagnóstico de MW.
Relativamente às estratégias terapêuticas, apesar de à época (2009), a opção ter recaído inicialmente no esquema CVP e depois R-CHOP, sabemos que atualmente esta última já não é considerada primeira linha de tratamento. As recomendações mais recentes baseiam-se na associação rituximab-bandamustina ou rituximab-bortezumib, este último se o doente apresentar características de alto risco como síndrome de hiperviscosidade14.
Neste contexto, os fatores de prognóstico associados a pior sobrevivência na MW e que faziam antever um prognóstico desfavorável no caso deste doente seriam a idade superior a 65 anos, a hemoglobina inferior ou igual a 11.5 g/dL, a contagem de plaquetas inferior ou igual a 100 x 109/L, a β2- microglobulina superior a 3 mg/L e a concentração de proteína monoclonal no soro superior a 7.0 g/dL9.
O tamanho da paraproteína IgM resulta em pouca excreção renal e apenas 20 % dos doentes excretam cadeias leves. Por isso a doença renal é incomum. Apesar deste facto, o doente apresentava uma insuficiência renal crónica, cuja etiologia não foi estudada e em que poderia ser considerada a hipótese de depósito de paraproteina a nível renal, que apenas seria detetada por biópsia renal.
Em conclusão, o presente caso de MW é ilustrativo de uma doença rara com uma apresentação inespecífica e insidiosa, que levou à necessidade de uma investigação clínica completa para reformulação de hipóteses clínicas até chegar a um diagnóstico definitivo.
Quadro I
Valores das análises sanguíneas realizadas nos dois internamentos (08/08/09 e 16/08/09) e em análises de rotina pedidas pelo médico assistente (28/01/09).
| Valores | 28-01-2009 | 08-08-2009 | 16-08-2009 |
| Hemoglobina (g/dL) | 9.9 | 5.5 | 6,4 |
| VGM (fL) | 94.9 | 93.2 | 94.4 |
| HGM (pg) | 33.5 | 32.1 | 31.6 |
| Leucócitos (céls/μL) | 2700 | 3500 | 7800 |
| Neutrófilos (céls/μL) | 1.400 | 2300 | 2500 |
| Linfócitos (céls/μL) | 1000 | 900 | 5100 |
| Plaquetas (céls/μL) | 113000 | 170000 | 123000 |
| Ureia (mg/dL) | - | 91 | 132 |
| Creatinina (mg/dL) | 1.7 | 2.1 | 3.3 |
| Bil total (mg/dL) | 1.85 | 0.20 | 6.28 |
| Bil directa (mg/dL) | 0.80 | <0.05 | 1.52 |
| LDH (U/L) | - | 417 | 2982 |
| β2 microglobulina | - | - | 7.11 |
Quadro II
Doseamento das proteínas séricas - segundo internamento
| Componente | Valor (intervalo normal) |
| Proteínas totais (g/dL) | 10,6 (6,3-8,2) |
| Albumina (%) | 39,5 (59,8-72,4) |
| α1 (%) | 2,5 (1,0-3,2) |
| α2 (%) | 5,4 (7,4-12,6) |
| β (%) | 49,3 (7,5-12,9) |
| γ (%) | 3,3 (8-15,8) |
| IgG (mg/dL) | 209 (700-1600) |
| IgA (mg/dL) | <25 (70-400) |
| IgM (mg/dL) | 7110 (40-230) |
| Cadeias Kappa (mg/dL) | 999 (130-370) |
| Cadeias Lambda (mg/dL) | 20 (90-210) |
| Relação κ/λ | 50 |
| Cadeias livres κ (mg/dL) | 134 (3.3-19.40) |
| Cadeias livres λ (mg/dL) | 8.71 (5,71-26,3) |
| Relação κ/λ | 15,38 (0,26-1,65) |
Quadro III
Imunofenotipagem da biópsia de medula óssea
| Componente | ||||
| Células T | CD3:14% | CD4: 6% | CD8: 7% | CD4/CD8: 0.86 |
| Células NK | CD3 - | CD16 + | CD56 +: | |
| Células B | CD19: 7%; CD20 +; CD23 -/+; CD25 + | slg Kappa: + ; CD10 -; CD79b +; CD11c - | slg Lambda: -; BCL2: normal; CD38 -/+ | CD5: +; FMC7 -; CD103 - |
| Plasmócitos | cKappa +; CD38++ | cLambda - | CD19-/+ | CD20- |
Figura I
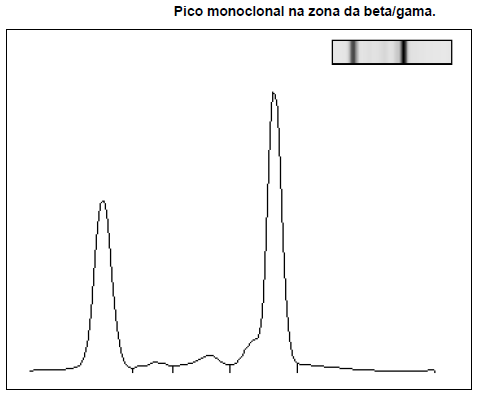
Eletroforese das proteínas plasmáticas segundo internamento
BIBLIOGRAFIA
BIBLIOGRAFIA
1. Fonseca R, Hayman S. Waldenström macroglobulinaemia. Br J Haematol. 2007 Sep;138(6):700-20.
2.Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, et al. The World Health Organization classification of neoplastic diseases of the haematopoietic and lymphoid tissues: Report of the Clinical Advisory Committee Meeting, Airlie House, Virginia, November 1997, Histopathology 2000; 36 (1): 6986
3. Herrinton LJ, Weiss NS. Incidence of Waldenstroms macroglobulinemia, Blood 1993; 82, 31483150.
4. Groves FD, Travis LB, Devesa SS, Ries LA, Fraumeni Jr JF. Waldenstroms macroglobulinemia: incidence patterns in the United States, 19881994, Cancer 1998; 82, 10781081.
5. Treon SP, Hunter ZR, Aggarwal A, et al. Characterization of familial Waldenstroms macroglobulinemia. Ann Oncol. 2006;17(3):488-494.
6. Elsawa SF, Novak AJ, Ziesmer SC, et al: Comprehensive analysis of tumor microenvironment cytokines in Waldenstrom macroglobulinemia identifies CCL5 as a novel modulator of IL-6 activity. Blood 2011, 118:5540-5549,
7. Treon SP, Xu L, Yang G, et al: MYD88 L265P somatic mutation in Waldenstroms macroglobulinemia. N Engl J Med 2012, 367:826-833
8. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014, May 1;123(18):2791-6.
9. Morel P, Duhamel A, Gobbi P, Dimopoulos MA, Dhodapkar MV, McCoy J, et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood 2009;113(18):4163-70.
10. Dimopoulos MA, Panayiotidis P, Moulopoulos LA, Sfikakis P & Dalakas M Waldenstroms macroglobulinemia: clinical features, complications, and management [Review], Journal of Clinical Oncology 2000; 18, 214226.
11. Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstroms macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstroms Macroglobulinemia, Semin Oncol 2003; 30:110-115
12. Kyle RA, Therneau TM, Rajkumar SV, et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance, Blood 2003;102:3759-3765
13. Kyle RA, Treon SP, Alexanian R, et al. Prognostic markers and criteria to initiate therapy in Waldenstroms macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstroms Macroglobulinemia, Semin Oncol 2003; 30:116-120
14. Dimopoulos MA, Kastritis E, Owen RG, et al. Treatment recommendations for patients with Waldenström macroglobulinemia (WM) and related disorders: IWWM-7 consensus. Blood 2014, 124: 1404-1411
15. San Miguel JF, Vidriales MB, Oci OE, et al. Immunophenotypic analysis of Waldenstroms macroglobulinemia, Semin Oncol 2003; 30 187-195