

Introdução:
A síndrome hepatopulmonar (SHP) é definida pela tríade de doença hepática, alterações da oxigenação arterial (gradiente alvéolo-arterial de oxigénio (P(A-a)O2) ≥15mmHg ou > 20 mmHg se idade > 64 anos) associada ou não a hipoxemia (pressão arterial de oxigénio (PaO2) a ar ambiente <80 mmHg) e evidência de vasodilatação intrapulmonar1. A sua prevalência varia entre 4 e 30% nos indivíduos com cirrose, dependendo da população estudada e dos critérios utilizados1-3. Uma task force da European Respiratory Society propôs uma classificação que utiliza a PaO2 para avaliar a gravidade da SHP em ligeira (PaO2≥ 80 mmHg), moderada (PaO2 ≥ 60 e <80 mmHg), grave (PaO2 ≥ 50 mmHg e <60 mmHg) e muito grave (PaO2 <50 mmHg)4. O prognóstico está diretamente relacionado com o seu estadio. Na maioria dos estudos, a presença ou gravidade do SHP não parece correlacionar-se com a gravidade da doença hepática crónica, avaliada através dos scores de Child-Turcotte-Pugh ou MELD (Model for End-stage Liver Disease)2,3,5.
A patogénese da SHP é complexa e não totalmente compreendida. O conhecimento atual advém maioritariamente de estudos experimentais em modelos animais. A principal alteração estrutural pulmonar é a dilatação vascular anormal (50-80 µm; intervalo normal 8-15 µm), desde o nível pré-capilar até ao pós-capilar, causando uma alteração da ventilação-perfusão e hipoxemia arterial. Isto permite que o sangue venoso misto alcance as veias pulmonares precocemente. Por outro lado, os vasos arteriovenosos colaterais (shunt direito-esquerdo) podem contribuir como bypass ao alvéolo, agravando ainda mais a hipoxemia1-3. Adicionalmente, o estado circulatório hiperdinâmico observado na cirrose hepática conduz a uma diminuição do tempo de hematose, isto é, o tempo necessário para a passagem dos eritrócitos ao nível da unidade alvéolo-capilar, comprometendo a difusão de oxigénio. A produção excessiva de óxido nítrico (ON) a nível da microcirculação pulmonar desempenha um papel central na patogénese da SHP. A relação com a insuficiência hepática e/ou hipertensão portal parece estabelecer-se, por um lado, através da produção hepática excessiva de endotelina 1 e, por outro, pela sobreprodução de fator de necrose tumoral alfa, resultado da translocação bacteriana e endotoxemia. Aquelas substâncias atuam por aumento da atividade das sintases endotelial e indutível do ON, respetivamente5-8. Estudos experimentais têm levantado a hipótese da importância de outras substâncias vasodilatadoras nos mecanismos da SHP, incluindo a angiogénese3,5-6.
A presença de sintomas ou sinais é insuficiente para a identificação desta síndrome, pelo que é necessário uma estratégia de pesquisa sistemática deste diagnóstico, particularmente em doentes com cirrose hepática7. A SHP é geralmente diagnosticada na sexta década de vida, sem consistente associação com o sexo ou etiologia da doença hepática3. A dificuldade de diagnóstico desta síndrome relaciona-se com a sua sintomatologia inespecífica. A maioria dos doentes apresenta dispneia associada à presença de estigmas de doença hepática crónica ao exame físico1-3,5-7. A dispneia, com um início insidioso e progressivo, particularmente com o exercício, é frequente na evolução natural da cirrose e só em alguns casos está relacionada com a SHP. Em contrapartida, a platipneia (dispneia agravada pelo ortostatismo e aliviada pelo decúbito) e a ortodeoxia (exacerbação da hipoxemia na passagem da posição de decúbito para o ortostatismo) são as manifestações mais características e também se observam frequentemente na SHP5-7. A hipoxemia associada a alcalose respiratória é um achado frequente na cirrose, pelo que se torna fundamental calcular o gradiente alvéolo-arterial de oxigénio e comprovar a presença de ortodeoxia6. Se ambos os valores são normais descarta-se a SHP. Assim, é necessária uma estratégia de pesquisa sistemática deste diagnóstico, dado que a presença de sintomas ou sinais é insuficiente para a identificação desta síndrome, particularmente em doentes com cirrose hepática8.
Os autores descrevem de seguida o caso clínico de uma doente com SHP no contexto de infeção VIH e cirrose hepática e revêem aspetos fundamentais do seu diagnóstico e abordagem.
Caso Clínico
Uma doente do sexo feminino, de 56 anos de idade, foi referenciada à consulta de Medicina Interna em março de 2007 por infeção pelo VIH-1 reconhecida cerca de 2 meses antes, adquirida possivelmente por comportamento sexual de risco. Tinha história de hábitos etílicos marcados e possível doença hepática crónica.
Na primeira avaliação referia dispneia para pequenos esforços e tosse persistente. À observação, realçavam-se telangiectasias faciais, cianose labial, sequelas de infeção por herpes zoster no dorso e hipocratismo digital. Apresentava taquipneia em repouso (28 ciclos/min). A saturação arterial periférica (SatO2) a ar ambiente por oximetria de pulso era de 77%. Na auscultação pulmonar salientava-se diminuição global do murmúrio vesicular e a auscultação cardíaca era normal. A nível abdominal, esplenomegalia, sem circulação colateral ou ascite. Dos exames complementares iniciais destacavam-se: contagem de linfócitos T CD4+ 226/mm3, carga viral (ARN do VIH) 244000 cópias/ml; hemoglobina 14.8 g/dl, plaquetas 104.000/mm3, alanina transaminase 50 U/L, desidrogenase lática 576 U/L e albumina 2.2 g/dl. A bilirrubina total era normal. O estudo da coagulação mostrou prolongamento do tempo de protrombina e INR (respetivamente, 18.5 segundos e 1.55). Na telerradiografia de tórax, presença de infiltrado reticulo-intersticial de predomínio nos lobos inferiores. A avaliação gasométrica mostrava insuficiência respiratória hipoxémica e aumento do gradiente alvéolo-arterial de O2 (PaO2 44 mmHg, PaCO2 31 mmHg; P(A-a)O2 42 mmHg).
Em Janeiro de 2008 iniciou terapêutica anti-retrovírica (efavirenz, tenofovir e emtricitabina), com supressão da replicação vírica, mas sem recuperação da imunidade (CD4+ 150-200/mm3). A doente referia agora dispneia mesmo em repouso e verificava-se poliglobulia secundária à hipoxemia, apesar da oxigenioterapia suplementar e da realização de flebotomias de forma regular. A prova de função respiratória revelou espirometria normal e capacidade de difusão de monóxido de carbono (DLCO) diminuída (63%). O ecocardiograma transtorácico foi normal (boa função sistólica, sem valvulopatias, derrame pericárdico ou shunt intra-cardíaco), permitindo calcular a pressão sistólica da artéria pulmonar (PSAP) em 25 mmHg. A tomografia computorizada do tórax (incluindo cortes de alta resolução) não tinha alterações.
Perante uma doente com sintomas pulmonares e hipoxemia em que tinha sido excluída patologia cardiopulmonar significativa, foi colocada a hipótese de as alterações pulmonares se enquadrarem na patologia hepática de base. O ecocardiograma transtorácico com contraste evidenciou o aparecimento de microbolhas a nível das cavidades cardíacas esquerdas após cinco ciclos cardíacos, sugerindo a presença de vasodilatação pulmonar. A cintigrafia de perfusão pulmonar com agregados de albumina marcados com tecnésio-99 (Tc-99) permitiu quantificar a fração de shunt intra-pulmonar em 21% (figura 1). Estes resultados, face ao hipocratismo digital associado a hipoxemia grave em doente com doença hepática e sem patologia cardiopulmonar significativa, confirmaram a hipótese diagnóstica de síndrome hepatopulmonar.
No decurso de 2010 observou-se melhoria da tolerância ao esforço e necessidade de oxigenioterapia suplementar apenas no período noturno. Entretanto foi referenciada à consulta de Hepatologia. Da investigação etiológica do quadro de doença hepática crónica salientavam-se: serologia negativa para hepatite B e C; ferro, ferritina, transferrina, capacidade total de fixação de ferro, imunoglobulinas e função tiroideia sem alterações; alfa 1-antitripsina, ceruloplasmina e alfa-fetoproteína (AFP) dentro dos valores de referência e auto-imunidade (anticorpos anti-nucleares, anti-ADN, anti-músculo liso e anti-mitocôndria) negativa. A ecografia abdominal revelou fígado com dimensão normal e ecoestrutura heterogénea e de contornos “rombos”, esplenomegalia (12 cm) e ausência de ascite. A endoscopia digestiva alta mostrou varizes grau I no terço médio e inferior do esófago. Foram adicionadas à medicação habitual furosemida e propranolol. A biópsia hepática por via transjugular, com avaliação hemodinâmica da circulação portal, permitiu calcular o gradiente portal de 17 mmHg, confirmando a presença de hipertensão portal. O exame histológico mostrou a presença de septação fibrosa e transformação nodular do parênquima, já cirrose, e nos espaços porta infiltrado inflamatório mononucleado com granulomas epitelióides, sem necrose nem células gigantes mononucleadas, e na interface epitélio-mesenquimatosa lesões necro-inflamatórias crónicas.
No fim de 2010 verificou-se elevação marcada da AFP para 1251 ng/ml (valor normal até 13.4 ng/ml). A ecografia hepática com doppler e, posteriormente, a tomografia computadorizada abdominal com contraste mostraram a presença de lesão sólida a nível da transição dos segmentos hepáticos V e VI, com 7,7 cm de maior diâmetro, compatível com carcinoma hepatocelular, associada a trombose do ramo direito da veia porta. Mais tarde, a arteriografia hepática identificou lesão tumoral infiltrativa no lobo direito e fistulização arterio-portal por invasão tumoral e persistência de trombo endoluminal a nível do ramo direito da veia porta. Observou-se agravamento clínico progressivo, com encefalopatia hepática, icterícia e ascite. Propostos os melhores cuidados de suporte, dada a condição clínica e o prognóstico reservado a curto prazo da doente. A doente faleceu em junho de 2011 com peritonite bacteriana espontânea.
Discussão:
A SHP é uma complicação importante da doença hepática crónica. A hipótese diagnóstica da síndrome hepatopulmonar deve ser equacionada em qualquer doente com doença hepática crónica que desenvolve dispneia ou hipoxemia8. A investigação etiológica da hipoxemia consiste em determinar se existe patologia pulmonar, parenquimatosa ou não, e/ou complicações da própria patologia de base2,5.
O ecocardiograma transtorácico contrastado através da injeção por veia periférica de soro salino agitado para produzir microbolhas (≥20 µm de diâmetro) é o exame mais sensível e menos invasivo que permite fundamentar a suspeita de dilatação vascular pulmonar. Normalmente, as microbolhas são conduzidas das cavidades direitas do coração para a circulação pulmonar onde são absorvidas e não atingem as cavidades esquerdas. A observação das microbolhas nas cavidades esquerdas antes do terceiro ciclo indica a presença de shunt intracardíaco direito-esquerdo. No caso de shunt intra-pulmonar as microbolhas são observadas apenas ao fim de três a seis ciclos cardíacos2,3.
Outro método menos usado para o diagnóstico é a cintigrafia de perfusão pulmonar com macroagregados de albumina marcados com produto radioativo (tecnésio-99m), que permite quantificar o grau de vasodilatação intra-pulmonar (fração de shunt). Em caso de shunt direito-esquerdo observa-se captação do produto marcado superior a 6% a nível sistémico (renal, cerebral e tecidos moles), acima do que ocorre em indivíduos saudáveis. No entanto, não permite diferenciar se o shunt é cardíaco ou pulmonar e possui menor sensibilidade que o ecocardiograma transtorácico com contraste5-7.
Em circunstâncias particulares, a angiografia pulmonar também pode ser usada para complementar o diagnóstico de SHP, nomeadamente naqueles doentes com hipoxemia grave (PaO2 <60 mmHg), com escassa resposta a 100% de oxigénio (aumento na PaO2 para menos de 300 mmHg) e se houver forte suspeição da existência de comunicação arteriovenosa que possa ser resolvida por embolização6,7. Até ao momento não existe terapêutica médica específica para o SHP. A suplementação de oxigénio em doentes com PaO2 <60 mmHg ou com dessaturação induzida pelo exercício pode corrigir a hipoxemia, embora sem melhoria na sobrevida global1-3,6. O transplante hepático é o tratamento mais eficaz. A SHP está associada a redução da sobrevida média (10.6 vs 40.8 meses), a aumento da mortalidade (75% vs 13.6%; p=0.017) dos doentes que aguardam transplante hepático, bem como a aumento da mortalidade após transplante. Na literatura estão descritas taxas de reversibilidade da SHP de aproximadamente 80% dos casos ao fim de 15 meses após o transplante; no entanto, no caso das formas mais severas de SHP, a morbimortalidade associada é superior9,10. Assim, no doente com SHP é fundamental referenciar precocemente para transplante hepático, dado que constitui indicação e tem prioridade na lista de espera.
A doente descrita tinha infeção pelo VIH, consumo crónico de álcool, interrompido após o diagnóstico, e cirrose hepática sem outras causas aparentes. As descrições de casos de SHP em doentes com VIH na literatura são pontuais, referentes a co-infetados com o vírus da hepatite C em fase de cirrose11,12. Neste caso verificou-se uma evolução mais indolente do que o esperado. De facto, a melhoria da oxigenação na doente foi intrigante: poder-se-á admitir que se tratou de evolução espontânea ou especular sobre a influência positiva da terapêutica anti-retrovírica. Em relação à abordagem terapêutica, a referenciação para transplante hepático foi ponderada. Contudo, inicialmente considerou-se que o risco de morbimortalidade pós-transplante era muito elevado, dada a gravidade da síndrome hepatopulmonar (PaO2 <50 mmHg e fração de shunt >20%). Posteriormente, face à melhoria da hipoxemia verificada e à persistência de importante imunossupressão, optou-se por manter abordagem conservadora. O diagnóstico de carcinoma hepatocelular no decurso da evolução clínica agravou ainda mais o prognóstico da doente.
O caso clínico apresentado reflete a história natural da cirrose hepática e as suas complicações, evidencia a desafiante marcha diagnóstica da SHP e aflora as questões que se podem colocar na sua abordagem no caso raro de infeção simultânea pelo VIH.
Figura I
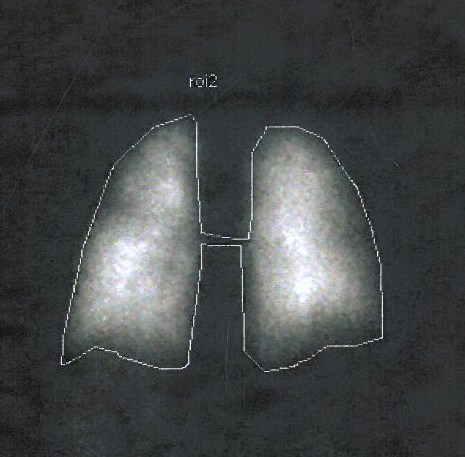
Cintigrafia de perfusão pulmonar. Captação pulmonar (A)
Figura II
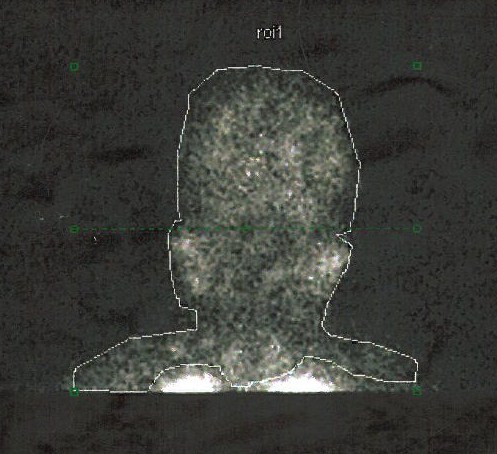
e extra-pulmonar (B) do radiofármaco.
BIBLIOGRAFIA
Bibliografia:
1. Varghese J, Ilias-basha H, et al. Hepatopulmonary syndrome – Past to present. Annals of Hepatology 2007; 6(3):135-142.
2. Rodrígues-Roisin R, Krowka MJ. Hepatopulmonary syndrome – A Liver-Induced Lung Vascular Disorder. N Engl J Med 2008; 358(22): 2378-2387.
3. Porres-Aguilar M, Altamirano JT, Torre-Delgadillo A, Charlton MR, Duarte-Rojo A. Pulmonary hypertension and hepatopulmonary syndrome: a clinician-oriented review. Eur Respir Rev 2012; 21 (125):223-233.
4. Rodríguez-Roisin R, Krowka MJ, Hervé P, Fallon MB. ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD). Eur Respir J 2004; 24:861-80.
5. Ho Vicent. Current concepts in the management of hepatopulmonary syndrome. Vascular Health and Risk Management 2008; 4(5):1035-1041.
6. Tumgor G. Cirrhosis and hepatopulmonary syndrome. World J Gastroenterol 2014; 20(10):2586-2594.
7. Palma DT, Fallon MB. The hepatopulmonary syndrome. Journal of Hepatology 2006;45:617-625.
8. Freire R, Mangualde J, Vieira AM, Lobato C, Alves LA, Cremers MI, Augusto F, Caetano F, Oliveira AP. Sindrome hepatopulmonar em doentes com cirrose hepática: importância da sua pesquisa sistemática e impacto no prognóstico. J Port Gastroenterol 2007;14:176-183.
9. Houlihan DD, Holt A, Elliot C, Ferguson JW. Review article: liver transplantation for the pulmonary disorders of portal hypertension. Liver Transplantation Aliment Pharmacol Ther 2013; 37: 183–194.
10. Swanson KL, Wiesner RH, Krowka MJ. Natural history of hepatopulmonary syndrome: impact of liver transplantation. Hepatol 2005;41:1122-1129.
11. Ferreira M, Gazzana M, Barreto S, Knorst M. Síndrome hepatopulmonar em paciente com cirrose por vírus C e SIDA. J Pneumol 2000; 27(1):52-55.
12. Torno M, Witt M, Sue D. Hepatopulmonary Syndrome in HIV-Hepatitis C Virus Coinfection: A Case Report and Review of the Literature. Clin Inf Dis 2004; 39: e25-9.