

Vasculite é um grupo heterogéneo de doenças caracterizado por uma inflamação da parede dos vasos sanguíneos, que dá origem a alterações no fluxo de sangue e, por vezes, a lesão da integridade estrutural do próprio vaso.(3) Dessa forma, podem resultar alterações estenóticas, oclusivas e/ou aneurismáticas.(4) A maioria das vasculites são idiopáticas ou primárias (3), embora algumas possam surgir no decurso de doenças já estabelecidas, como é o caso da vasculite associada a infeções ou medicamentos, a vasculite no lupus eritematoso sistémico (LES) e vasculite reumatoide.(2)
Afetam um ou vários órgãos e vasos de um ou mais tipos. A clínica sobrevém da isquemia dos tecidos nutridos pelos vasos lesionados ou da hemorragia associada à rotura de aneurismas (9). Os sintomas sistémicos de febre, perda ponderal, anorexia e sudorese (3) surgem muitas vezes a acompanhar o processo inflamatório generalizado. (6)
As vasculites podem consistir num processo relativamente benigno, loco-regional, como acontece na vasculite leucocitoclástica, ou sistémico e ameaçador de vida, como na vasculite associada a anticorpos anti-citoplasma dos neutrófilos (ANCA).
Há já algum tempo que se tem adotado a nomenclatura definida na Chapel Hill Consensus Conference para as vasculites sistémicas, que pretendeu reunir consenso para o nome a dar à maioria das vasculites e construir uma definição para cada.(7) Tem sido vastamente utilizada, embora não seja um sistema de classificação nem de diagnóstico (1, 7, 8) (Figura I).
Apesar dos progressos auferidos nas últimas décadas, as vasculites são doenças complexas, onde por vezes é difícil chegar a um diagnóstico definitivo pela variabilidade e sobreposição de achados clínicos que se podem encontrar.
Os autores apresentam o caso clínico de uma doente cujas manifestações, resultado de exames complementares de diagnóstico e evolução, fizerem repensar o diagnóstico inicialmente considerado, concluindo pelo de uma vasculite associada a ANCA (Granulomatose com poliangeíte - Wegener).
Caso clínicoMulher de 80 anos de idade, raça caucasiana, internada através por quadro febril arrastado. Tinha febre com um mês de evolução, sem predomínio de horário, com temperaturas axilares no domicílio que oscilavam entre os 38-39ºC.
Foi medicada com amoxicilina e ácido clavulânico para uma otite média 10 dias antes e, posteriormente, com levofloxacina e anti-inflamatórios não esteroides para uma infeção respiratória sem, no entanto, referir melhoria sintomática.
Tinha tosse seca com 3 meses de evolução, astenia, anorexia e emagrecimento. Queixava-se de cefaleias intensas de predomínio frontal, claudicação mandibular e cervicalgia de características mecânicas.
Tinha antecedentes patológicos de dislipidemia e sinusite, estando apenas medicada com sinvastatina.
Não referia viagens recentes, alergias, contacto com animais ou com pessoas com sintomatologia semelhante.
Negava hábitos alcoólicos, tabágicos, consumo de outros medicamentos ou substâncias.
Os antecedentes patológicos familiares eram irrelevantes para a presente situação.
Apresentava-se consciente, orientada no tempo e no espaço, colaborante. A pele e as mucosas estavam discretamente pálidas.
Estava eupneica em repouso. A tensão arterial era 106/65mmHg, temperatura auricular 37,9ºC, saturação periférica 98% em ar ambiente. Não tinha adenomegálias periféricas palpáveis.
A otoscopia era normal. A orofaringe não mostrava alterações. Não apresentava lesões oculares. Os pulsos temporais eram palpáveis e simétricos, de fraca intensidade.
A auscultação cardíaca revelou um sopro sistólico aórtico II/VI. A auscultação pulmonar era normal. Ao exame abdominal, apenas a palpação do flanco esquerdo se revelou discretamente dolorosa, sem serem evidentes massas ou organomegálias, ou sinais de irritação peritoneal.
Não tinha alterações dos membros, inflamação articular, nem lesões cutâneas.
O exame neurológico sumário, nomeadamente a força muscular, reflexos, pares cranianos e marcha, era normal. Não tinha rigidez da nuca.
Laboratorialmente constatou-se anemia normocrómica normocítica (hemoglobina de 11,3 g/dL), leucocitose (21,3x103/µL), neutrofilia (17,96x103/µL), eosinófilos normais (0,8x103/µL) e trombocitose (752x103/µL). A velocidade de sedimentação (VS) era elevada (99mm/h), tal como a proteína C reativa (PCR 221,60mg/L). Os restantes parâmetros analisados, entre os quais os valores de creatinina, ureia, enzimas hepáticas e hormonas tiroideias eram normais. A sumária de urina e o sedimento eram normais. A prova da tuberculina foi negativa.
Foi efetuado despiste de causas infeciosas e neoplásicas pelo que realizou culturas de líquidos biológicos, incluindo hemoculturas, que foram negativas. As serologias para os diferentes vírus e bactérias foram igualmente negativas ou não conclusivas. O proteinograma electroforético, a enzima de conversão da angiotensina (ECA) e marcadores tumorais nada revelaram que fizesse suspeitar o diagnóstico subjacente.
Também a ecografia abdominal, endoscopia digestiva alta, colonoscopia e ecocardiograma não mostraram alterações de relevo para a presente situação.
Já a radiografia torácica (figura 2) revelou um alargamento do mediastino superior e desvio da traqueia, confirmado pela tomografia computorizada (T.C.) torácica, pelo que se transcreve os dados mais importantes do relatório: ectasia do tronco e ramos da artéria pulmonar, alargamento do mediastino superior e ectasia da artéria pulmonar direita
O estudo da auto-imunidade foi negativo para os anticorpos anti-nucleares (ANA), mas positivo para os ANCA, subtipo mieloperoxidase (MPO ANCA > 600 U/mL).
Foi pedida T.C. dos seios que mostrou: hipertrofia da mucosa dos seios maxilares, labirinto etmoidal e seios frontais, processo inflamatório até às regiões osteo-infundibulares e recessos esfeno-etmoidais . A T.C. das órbitas foi normal.
O exame anatomopatológico da biópsia da artéria temporal direita foi compatível com vasculite granulomatosa (figura 3).
A doente foi medicada com prednisolona (1mg/Kg/dia) e encaminhada para a consulta externa de medicina.
Permaneceu clinicamente estável durante os primeiros 2 meses de tratamento. A VS baixou para 19 mm/1ªh e a PCR para 1,69 mg/L.
Deu novamente entrada na urgência por quadro de instalação súbita de febre, dispneia e astenia. A saturação periférica era 60%, temperatura auricular 38,1ºC, tensão arterial 98/56mmHg. Estava taquicardica e apresentava fervores bilaterais à auscultação pulmonar.
Fez radiografia torácica que revelou um infiltrado algodonoso bilateral (figura 4) e foi pedida angio - T.C. torácica onde se observaram extensas áreas de densificação parenquimatosa e sinais de tromboembolia pulmonar do ramo lobar médio, inferior direito e ramos segmentares (figura 5).
Iniciou pulsos de corticóides (1g/dia), ciclofosfamida 150mg/dia e enoxaparina 1mg/Kg/peso de 12/12h.
Por agravamento do quadro de insuficiência respiratória, foi transferida para o serviço de medicina intensiva, sob ventilação mecânica invasiva, tenho cumprido antibioterapia empírica (mas culturas negativas de líquidos biológicos). Foi extubada ao fim de alguns dias. Permaneceu em ventilação espontânea apenas 48h, após as quais veio a falecer.
DiscussãoDescreveu-se o caso de uma doente de 80 anos, com uma clínica que nos sugeria uma arterite de células gigantes, após se ter descartado a presença de doenças infeciosas ou neoplásicas subjacentes. O resultado histológico da biópsia efetuada à artéria temporal e os dados imagiológicos eram concordantes com o diagnóstico.
Esta vasculite é rara antes dos 50 anos e tem predileção pelos ramos extracranianos da artéria carótida, sendo a temporal frequentemente afetada. (2) As manifestações principais, que se podem sobrepor à síndroma da polimialgia reumática, são as constitucionais, as cefaleias e as outras alterações decorrentes do envolvimento das artérias do arco aórtico: diminuição da visão e claudicação da mandíbula. A biópsia da artéria temporal é a base do diagnóstico. Não está associada a nenhum marcador de auto-imunidade de forma particular. (4)
No entanto, a presença de ANCA, as alterações no trato respiratório superior, o desenrolar posterior da situação que revelou uma doença agressiva, que envolvia sobretudo os pulmões e poupava os rins, veio estabelecer como diagnóstico mais provável o de granulomatose com poliangeíte ou de Wegener.
Esta é uma vasculite necrotizante que afeta de forma preferencial pequenos vasos (capilares, arteríolas e vénulas), embora isto não signifique que os grandes vasos não possam estar envolvidos.(2) Apesar de existirem formas de doença limitada e de curso indolente, sobretudo quando relacionadas com manifestações do trato aéreo superior como sinusite, epistáxis, otite média, mastoidite ou úlceras nasais (4); podem tornar-se sistémicas e envolver os pulmões e o rim, com glomerulonefrite(2). Associa-se fortemente à presença de ANCA anti-proteinase 3 (PR3 - ANCA)(1,2,4). A principal causa de morte advém da instalação da síndroma reno-vascular, com hemorragia alveolar e falência renal secundária a glomerulonefrite rapidamente progressiva.
Considera-se que o interesse deste caso advém do facto de que foi diagnosticada uma vasculite granulomatosa por biópsia da artéria temporal, que é um vaso de grande calibre, e que é basilar no diagnóstico da arterite de células gigantes, mas numa doente com alterações pulmonares e título elevado de MPO-ANCA no sangue. Segundo a literatura, a presença de alterações pulmonares ocorre em apenas 9% dos casos de arterite da temporal, sendo os ANCA negativos.(9) Ainda assim, os MPO ANCA não são característicos da poliangeíte granulomatosa, estando presentes em apenas 20% dos casos.(10) Os MPO-ANCA são mais frequentes noutras vasculites de pequenos vasos como a poliangeíte microscópica e síndroma de Churg-Strauss, que também podem causar uma síndroma reno-pulmonar. Mas, a primeira não possui granulomas e a segunda, apesar de ser uma vasculite granulomatosa, está associada a asma e eosinofilia)(2,7).
Considera-se a pesquisa de ANCA nos testes de auto-imunidade útil em todas as doenças multissistémicas ainda não diagnosticadas, mas só por si e, como podemos constatar, não definem qual a vasculite em causa. Podem ter particular interesse, sobretudo se associados a sintomas clássicos e quando não há possibilidade de obter uma amostra tecidular para análise(1). Colocam-nos de sobreaviso para a eventualidade de uma patologia grave e para a necessidade de implementar mediadas terapêuticas imunossupressoras sem demora.
Figura I
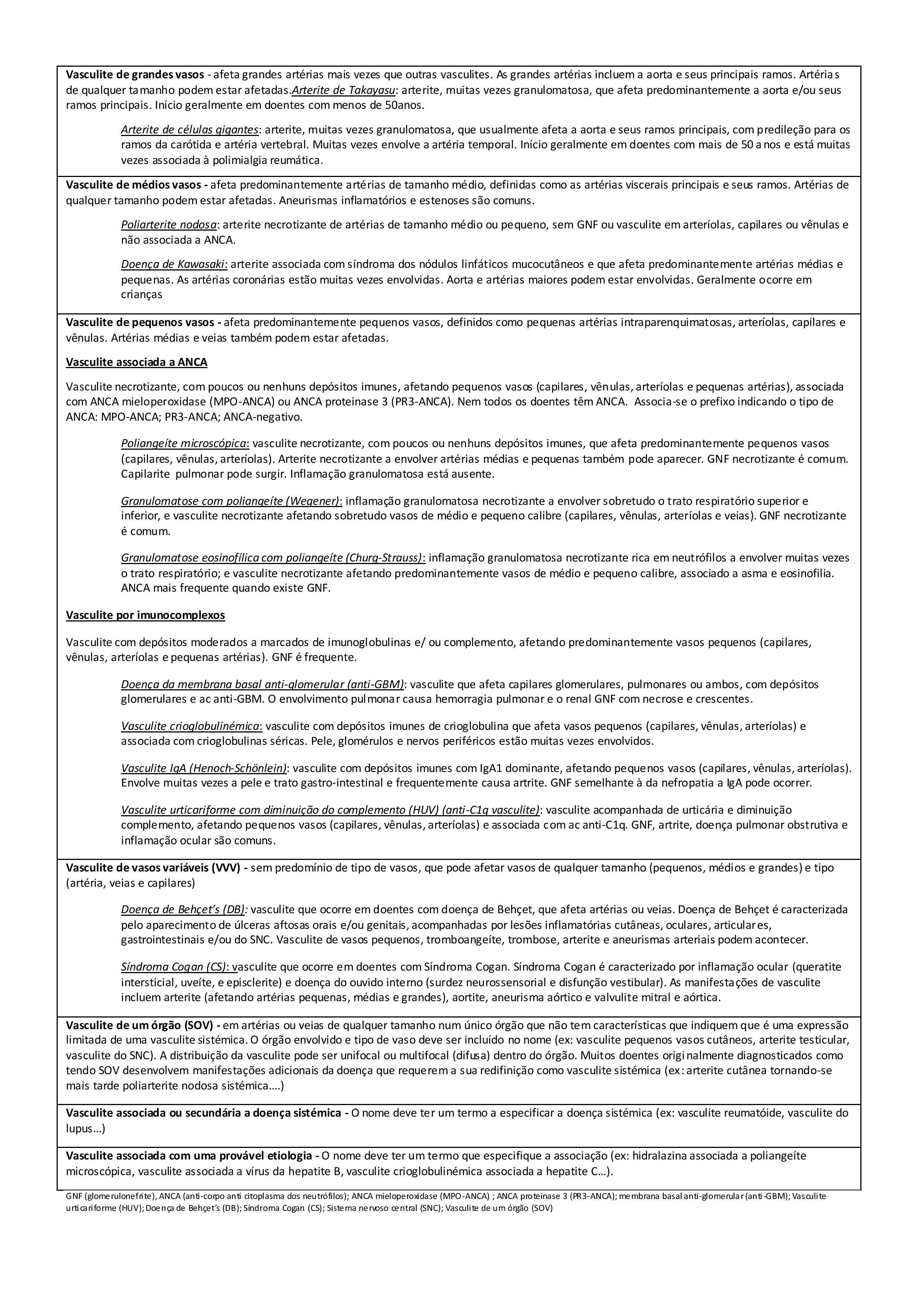
Nomenclatura e definição das vasculites , adaptado de Chapel Hill Consensus Conference 2012
Figura II
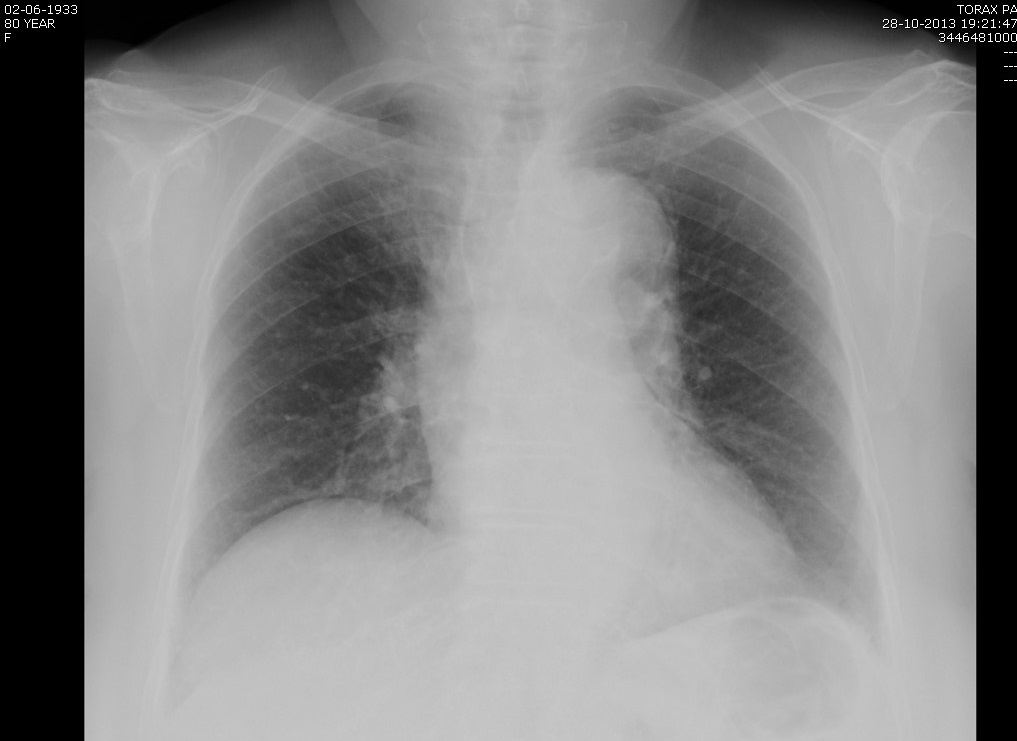
Radiografia torácica: alargamento do mediastino superior e desvio da traqueia
Figura III
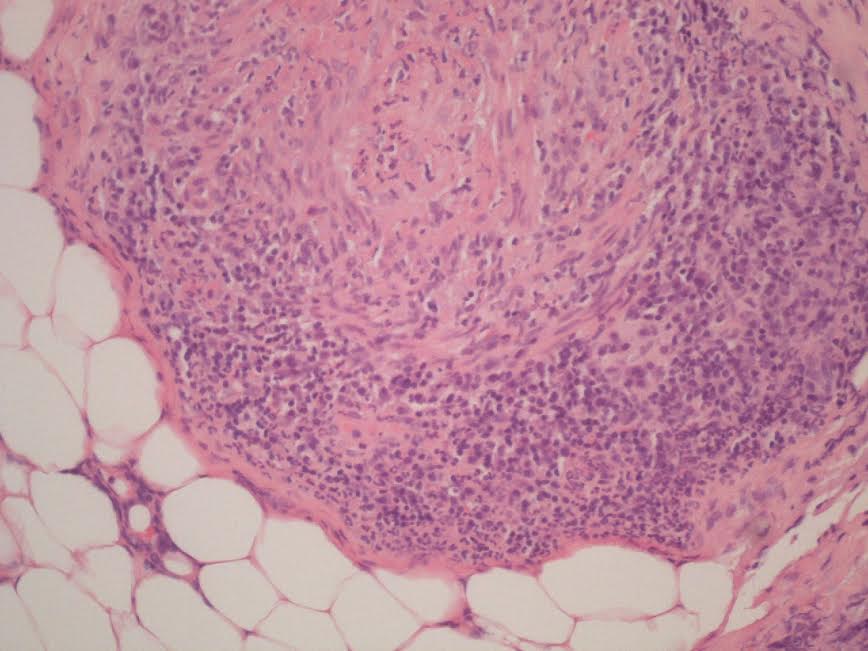
Biopsia da artéria temporal (HE 200x) - artéria muscular mostrando permeação intensa de toda a muscular própria e parte da adventícea por um componente inflamatório exuberante de predomínio mononucleado, onde ocasionalmente se observam alguns granulócitos e plasmócitos. Os agregados inflamatórios esboçam por vezes a formação de granuloma
Figura IV
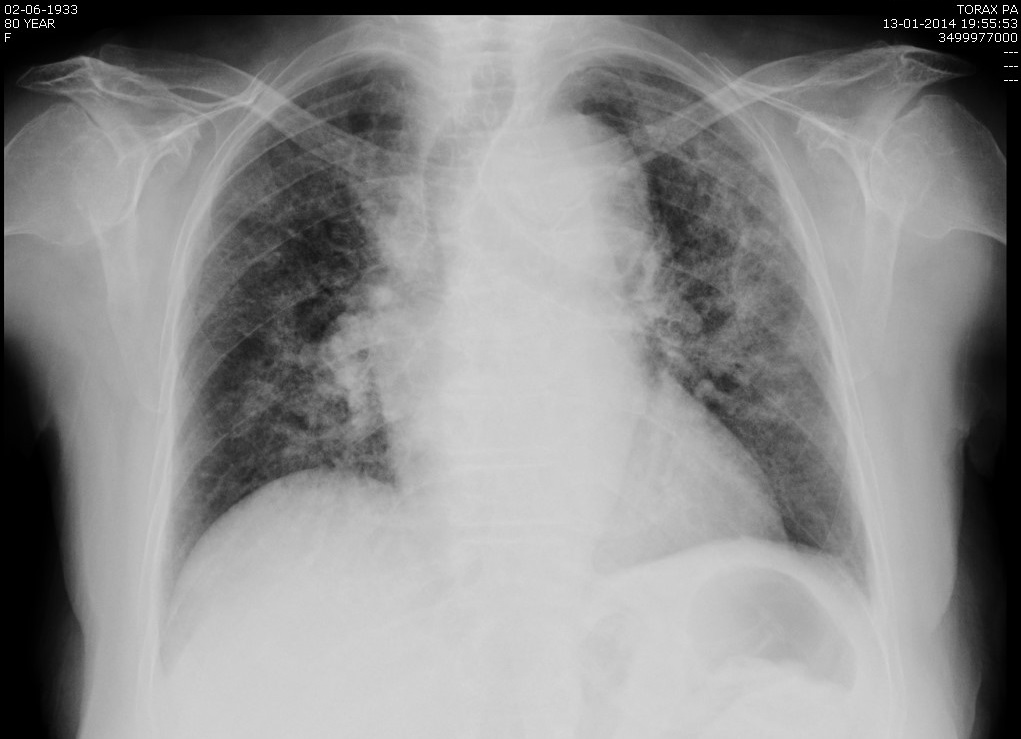
Radiografia torácica: infiltrado algodonoso bilateral
Figura V
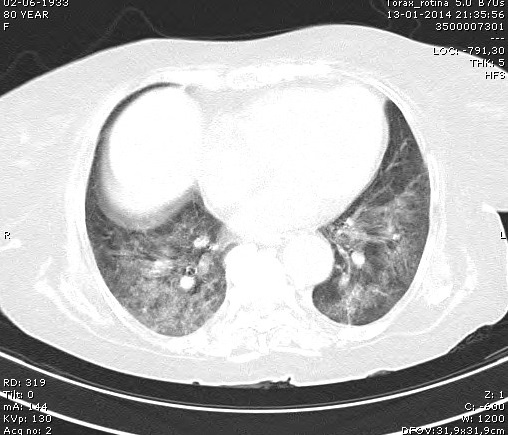
Angio-TAC
BIBLIOGRAFIA
1. Lugmani RA, Suppiah R, Gryson PC, Merkel PA, Watts R. Nomenclature and classification of vasculitis update on the ACR/Eular and classification of vasculitis study. Clin Exp immunol. 2011; 164: 11-13.
2. Harper L, Little MA. Small vessel vasculitides. Medicine 2009; 38:2: 84- 92.
3. Lightfoot Jr RW Jr. Overview of the inflammatory vascular diseases. In: Klippel JH, Dieppe PA. Rheumatology. London: Mosby, 1998; 7: 17.1-17.6.
4. Konttinem YT, Rotar Z, Pettersson T, Nordstrom DCE, Bacon P, Peterson J. Roadmap to vasculitis. Acta Reum Port. 2006; 3: 15-36.
5. Scott DGI, Watts RA. Epidemiology and clinical features of systemic vasculitis. Clin Exp Nephrol 2012; .doi: 10. 1007/s10157-013-0830-8
6. Watts R. Diagnosis and evaluation of vasculitis 2000. doi: 10.1093/rheumatology/ 39.3.245.
7. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F et all. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitis. Arthritis & Rheumatism 2013; 65: 1-11.
8. Lapraik C, Watts R, Bacon P, Carruthers D, Chakravarty K, DCruz D et all. BSR and BHPR guidelines for the management of adults with ANCA associated vasculitis. Rheumatology 2007; 46: 1-11.
9. Larson TS, Hall S, Hepper NGG, Hunder GG. Respiratory Tract Symptoms as a clue to Giant Cell Arteritis. Ann Intern Med. 1984; 101:594-597
10. Róman JJG; Hemorragias alveolares difusas pulmonares; Arch Bronconeumol.2008;44:428-36