

Introdução
A SAAF corresponde a uma alteração imunológica definida pela ocorrência de eventos trombóticos e/ou perdas fetais recorrentes na presença dos referidos anticorpos.1-2 Foi descrita pela primeira vez nos anos 80 do século XX por Hughes dando origem ao epónimo. 3
Os anticorpos antifosfolípidos (AAF) são imunoglobulinas que reagem com os fosfolípidos de carga negativa das membranas celulares. Dentro desta designação inclui-se o anticorpo anti-cardiolipina (ACL), o anticoagulante lúpico (AL) e o anticorpo anti-β2 glicoproteína I (anti-β2GPI).1-2 A presença de AAF positivos na população geral varia entre 1 e 5%, mas apenas uma minoria desenvolve SAAF.1 Estes podem estar aumentados isoladamente no contexto de outras patologias reumáticas ou auto-imunes, neoplasias, infeções virais, bacterianas ou parasitárias, anemia perniciosa e falciforme, diabetes mellitus, doença inflamatória intestinal, síndrome de Klinefelter, em doentes dialisados ou se existir uso concomitante de certos medicamentos.
Esta síndrome atinge igualmente o sexo masculino e feminino, sobretudo em idades jovens. 1
Quanto à sua classificação, pode ser primária, caso ocorra isoladamente, ou secundária, se coexistir com outras patologias autoimunes. Por exemplo, surge em cerca de 30% de doentes com lúpus eritematoso sistémico (LES).1 Uma derivação mais grave desta síndrome é conhecida como síndrome catastrófica dos anticorpos antifosfolípidos, que ocorre em menos de 1% dos doentes, e resulta de múltiplas oclusões vasculares acompanhadas de falência multiorgânica com elevada mortalidade.4
Caso clínico
Homem de 32 anos, caucasiano, enviado à consulta de Nefrologia por estudo analítico compatível com síndroma nefrótica, com proteinúria 7.7g/24h, colesterol total 336mg/dL, colesterol LDL 236mg/dL, colesterol HDL 52mg/dL, triglicéridos 238mg/dL e hipoalbuminemia (2.7g/dL).
Tinha sedimento urinário a documentar raros eritrócitos sem dismorfismo. Associadamente apresentava prolongamento do aPTT (86.8 segundos) e trombocitopenia (90000/µL), conhecida há dois anos.
O doente referia edema dos membros inferiores com cerca de um ano de evolução e agravamento progressivo ao longo do dia. Negava antecedentes prévios de eventos trombóticos, infeções recentes ou alterações macroscópicas da urina. Negava queixas articulares.
Dos antecedentes pessoais, de salientar dislipidemia sob atorvastatina 10mg id. Negava hábitos toxicómanos e alergias. Quanto aos antecedentes familiares, a mãe tinha histórico de abortos de repetição.
Ao exame objetivo evidenciava edema bimaleolar e sinal de Homans à direita. Sem sinais de livedo reticularis, isquemia digital, gangrena ou sinais de artrite nas mãos, cotovelos, ombros ou pés. Sem alterações mucocutâneas ou oculares, adenopatias palpáveis nas principais cadeias superficiais ou défices neurológicos. À auscultação cardíaca era evidente um sopro holossistólico, mais audível no foco mitral, de grau III/VI, sem irradiação. De salientar também hipertensão arterial (140/91mmHg). Sem febre.
Neste seguimento, foi pedido o estudo complementar. Para além das alterações analíticas referidas, o hemograma revelou velocidade de sedimentação aumentada (50mm/h). A bioquímica identificou ureia 60mg/dL, creatinina 1.30mg/dL e proteínas totais 5.3g/dL. O ionograma era normal. O eletrocardiograma não tinha sinais de isquemia aguda nem de hipertrofia ventricular esquerda nem a radiografia torácica sinais de derrame pleural ou traços de fibrose. Realizou um ecodoppler venoso dos membros inferiores que revelou trombose da veia poplítea direita. Dirigido à etiologia da síndroma nefrótica e por forte suspeita de uma causa secundária, foi realizado o estudo da autoimunidade. Assim, numa primeira fase, o anticorpo ACL foi positivo (IgG superior a 280.0GLPU/m e IgM 4.0MPLU/mL), tal como o anticorpo anti- β2GPI (IgG51.0U/mL e IgM 0.9U/mL) e o AL (sílica clotting time com razão 2.56). Associadamente, a pesquisa de fator V de Leiden, homocisteinemia, mutação do gene da protrombina e défice de antitrombina III foi negativa e as proteínas C e S estavam dentro dos valores da normalidade. Os ANAs foram positivos em três doseamentos. A restante autoimunidade foi negativa, nomeadamente os anticorpos anti-dsDNA, anti-SSA/Ro, anti-SSB/La, anti-RNP, anti-músculo estriado, anti-Sm, p-ANCA, c-ANCA, fração C3, C4 e CH50 do complemento, fator nefrítico C3 do complemento e anticorpo anti-receptor fosfolipase A2. A eletroforese de proteínas séricas tinha apenas a fração alfa 2 aumentada com 16.7% (Fig. 1). As serologias para o vírus da imunodeficiência humana, hepatite B e C foram não reativas. O teste de Coombs direto foi positivo (C3d+) na ausência de anemia hemolítica. O doseamento dos fatores de coagulação II, VII, VIII, IX e X também foi normal, à exceção da elevação do fator XIII (207.3%). O fator de von Willebrand não se encontrava diminuído. A ecografia não demonstrou alterações de relevo relativas à morfologia renal. Quanto à biópsia renal, realizada após administração de desmopressina intra-nasal e plasma fresco congelado, revelou, através de análise histopatológica por microscopia eletrónica, glomerulonefrite membranosa em estádio II com lesões de esclerose segmentar. A imunofluorescência demonstrou discretos depósitos granulosos de IgG nas paredes (Fig. 2). A Periodic acid-Schiff (PAS) surgiu com esclerose e paredes espessadas (Fig. 3) e o tricrómio com paredes espessadas e hipertrofia de podócitos (Fig. 4). Por outro lado, dado a existência de um sopro cardíaco, foi pedido um ecocardiograma transtorácico que descreveu a área mitral com aspeto mixomatoso. Este foi posteriormente complementado com ecocardiograma transesofágico que acrescentou pequenas lesões vegetantes compatíveis com endocardite de Libman-Sacks (Fig. 5). De referir que as hemoculturas realizadas previamente não identificaram nenhum agente.
Neste contexto, o diagnóstico considerado mais provável foi o de SAAF secundário a LES. Por esse motivo, o doente iniciou imunossupressão com micofenolato de mofetil e corticoesteróides, hipocoagulação com varfarina, assim como ramipril 5mg id. Num segundo doseamento, doze semanas após, confirmou-se apenas positividade do anticorpo anti- β2GPI (IgG 31.5U/mL e IgM 0.5U/mL) e do AL (sílica clotting time com razão 1.66). O ecocardiograma transesofágico mantinha vegetações ao nível da válvula mitral.
Posteriormente, por não haver resposta ao micofenolato de mofetil, este foi substituído por ciclosporina, com descida progressiva da proteinúria para valores de 4g/24h.
Discussão
A patogénese da SAAF não se encontra bem definida. Uma das hipóteses defende que a exposição a alguns agentes infeciosos induz a produção destes anticorpos em indivíduos susceptíveis ou no contexto de doenças reumáticas como o LES.1,5 No entanto, uma vez que um dos AAF esteja presente parece ser necessário um segundo evento para desenvolver a SAAF, tal como traumatismo, imobilização prolongada, gravidez e período pós-parto, uso de anticoncetivos orais, terapêutica hormonal de substituição, neoplasias, síndroma nefrótica ou dislipidemia.5 Considera-se haver uma predisposição genética através da mutação de certos fatores de coagulação.5
A manifestação clínica mais frequente da SAAF é a trombose venosa profunda seguida da trombocitopenia, livedo reticularis, acidente vascular cerebral, tromboflebite superficial, tromboembolismo pulmonar, perdas fetais, acidente isquémico transitório e anemia hemolítica.5-6
Assim, devemos suspeitar de SAAF perante um ou mais eventos trombóticos sem etiologia definida, se existir histórico de complicações relacionadas com a gravidez ou se analiticamente houver evidência de trombocitopenia ou prolongamento do aPTT não explicados por outro motivo, como demonstrado pelo caso clínico descrito.1-2,5-6
O diagnóstico diferencial integra outras causas de trombose venosa ou arterial, a migroangiopatia trombótica isolada e a iatrogenia medicamentosa.
Em relação ao diagnóstico definitivo, segundo os critérios de Sapporo revistos em 2004 e divulgados sob a forma de consensos em 2006, este é estabelecido na presença de pelo menos um critério laboratorial associado a um critério clínico.1-2,5 Dos critérios laboratoriais fazem parte o doseamento dos anticorpos ACL (IgG e IgM) e anti- β2GPI (IgG e IgM) por Enzyme-Linked Immunosorbent Assay (ELISA) e testes para AL (dilute Russell viper venom time e aPTT) que devem ser sempre repetidos após doze semanas.1-2,5 Os critérios clínicos incluem a morbilidade na gravidez (morte fetal após 10 semanas de gestação com morfologia fetal normal; parto prematuro por eclâmpsia/pré-eclâmpsia ou insuficiência placentária, antes das 34 semanas de gestação, com neonato normal; três ou mais abortos consecutivos antes das 10 semanas de gestação, não explicados por outro motivo) e a trombose vascular documentada (pequenos vasos, arterial ou venosa). 1-2 Atualmente existe alguma divergência quanto à definição de critérios clínicos. Há autores que defendem a inclusão de critérios considerados não clínicos na classificação da SAAF, tais como a trombocitopenia, a lesão valvular cardíaca e a nefropatia causada por esta síndrome. 6 Como fundamento, está sobejamente documentada a elevada prevalência de doença cardiovascular associada ao SAAF e LES. Sobretudo o caso particular da endocardite de Libman-Sacks que pode levar a fenómenos tromboembólicos e regurgitação ou estenose valvular, sendo que a lesão imunológica parece ter um papel central no seu desenvolvimento.7
Quanto às manifestações renais da SAAF, existe um amplo espectro de eventos trombóticos, tais como estenose da artéria renal, enfarte renal, trombose da veia renal e microangiopatia trombótica. As lesões vasculares intrarrenais também têm sido descritas, nomeadamente a nefropatia por AAF, cuja apresentação é variável. Os doentes podem permanecer assintomáticos com função renal normal e proteinúria inferior a 2g/24h ou então com lesão renal aguda e proteinúria compatível com síndroma nefrótica, hematúria esporádica e hipertensão arterial. A clínica reflete a diversidade de achados histológicos que incluem isquemia glomerular e lesões trombóticas sem depósitos imunes glomerulares ou arteriais na imunofluorescência. Cerca de um terço dos doentes apresentam lesões menos características, como doença de lesões mínimas, glomerulonefrite pauci-imune, glomeruloesclerose focal e segmentar e glomerulonefrite membranosa. 8-9 Em relação ao LES, surgiram evidências recentes que consideram a podocitopatia como uma entidade específica de nefrite lúpica. 10
Por fim, o tratamento da SAAF, para além das medidas de modificação de estilo de vida, baseia-se sobretudo na antiagregação plaquetária com ácido acetilsalicílico e na hipocoagulação com varfarina ou enoxaparina.11-12
Com as medidas terapêuticas apropriadas, a maior parte dos doentes tem um bom prognóstico a longo prazo. 1-2
Figura I
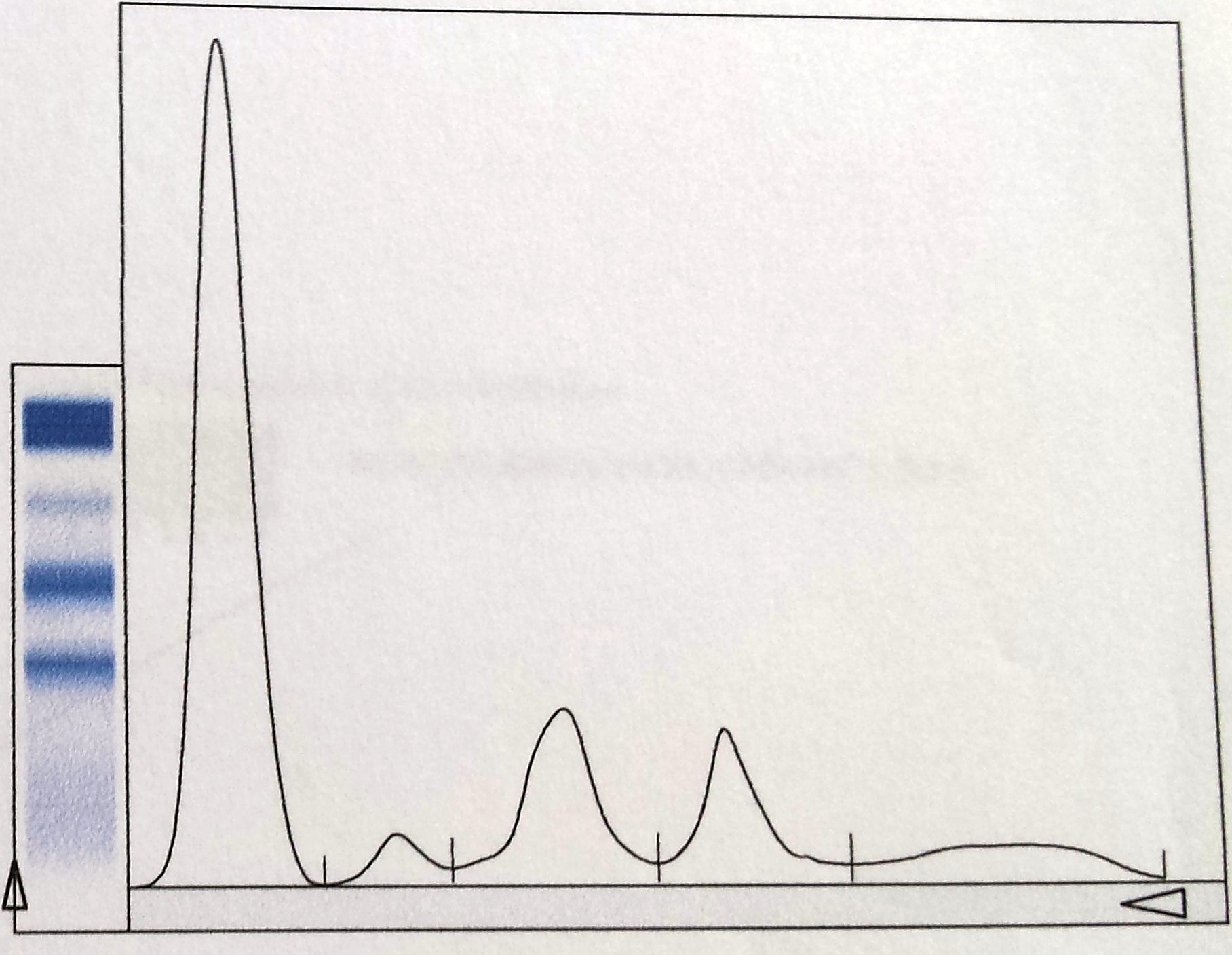
Eletroforese de proteínas séricas com fração alfa 2 aumentada (16.7%).
Figura II
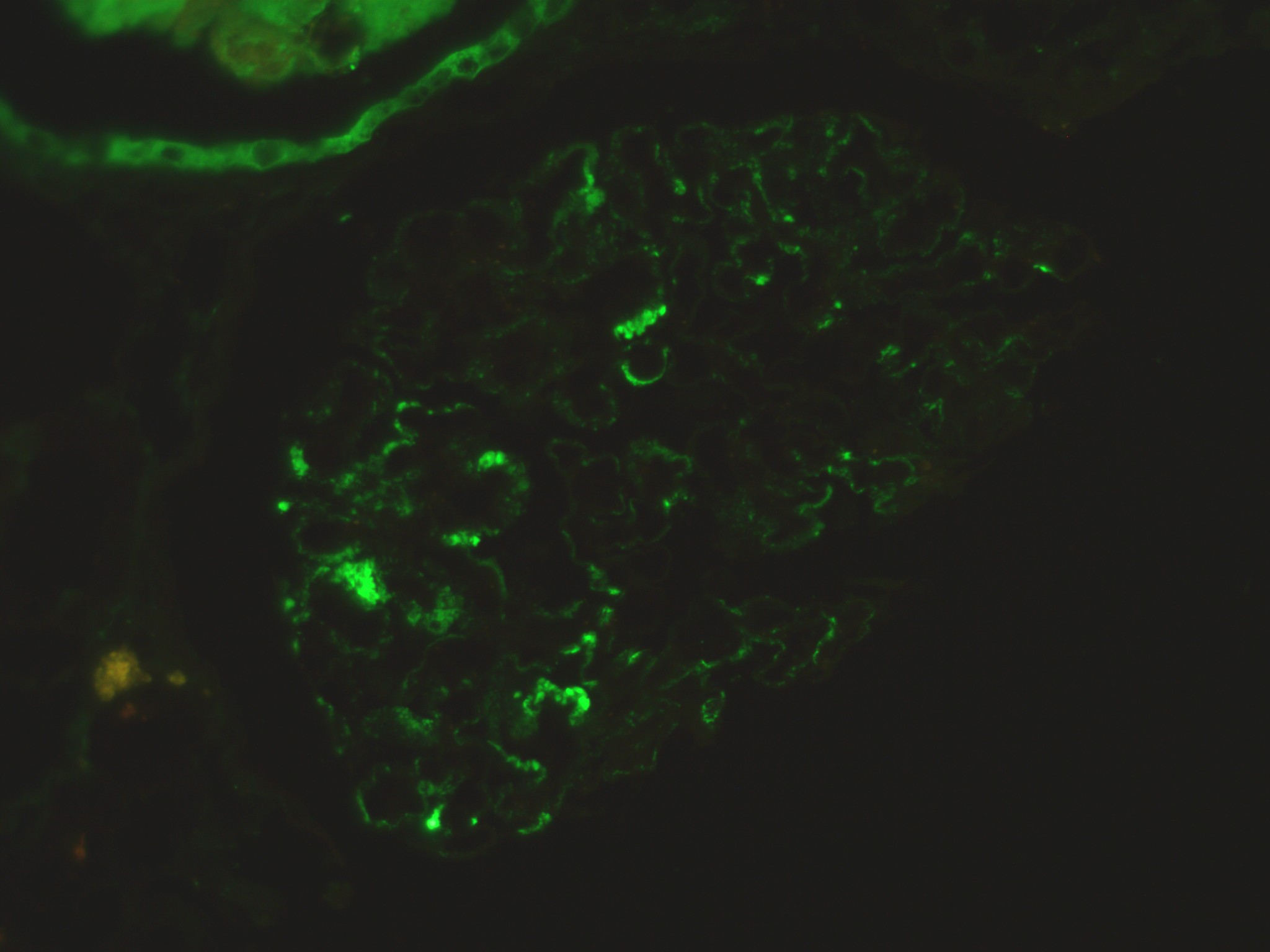
Imunofluorescência com discretos depósitos granulosos de IgG nas paredes.
Figura III
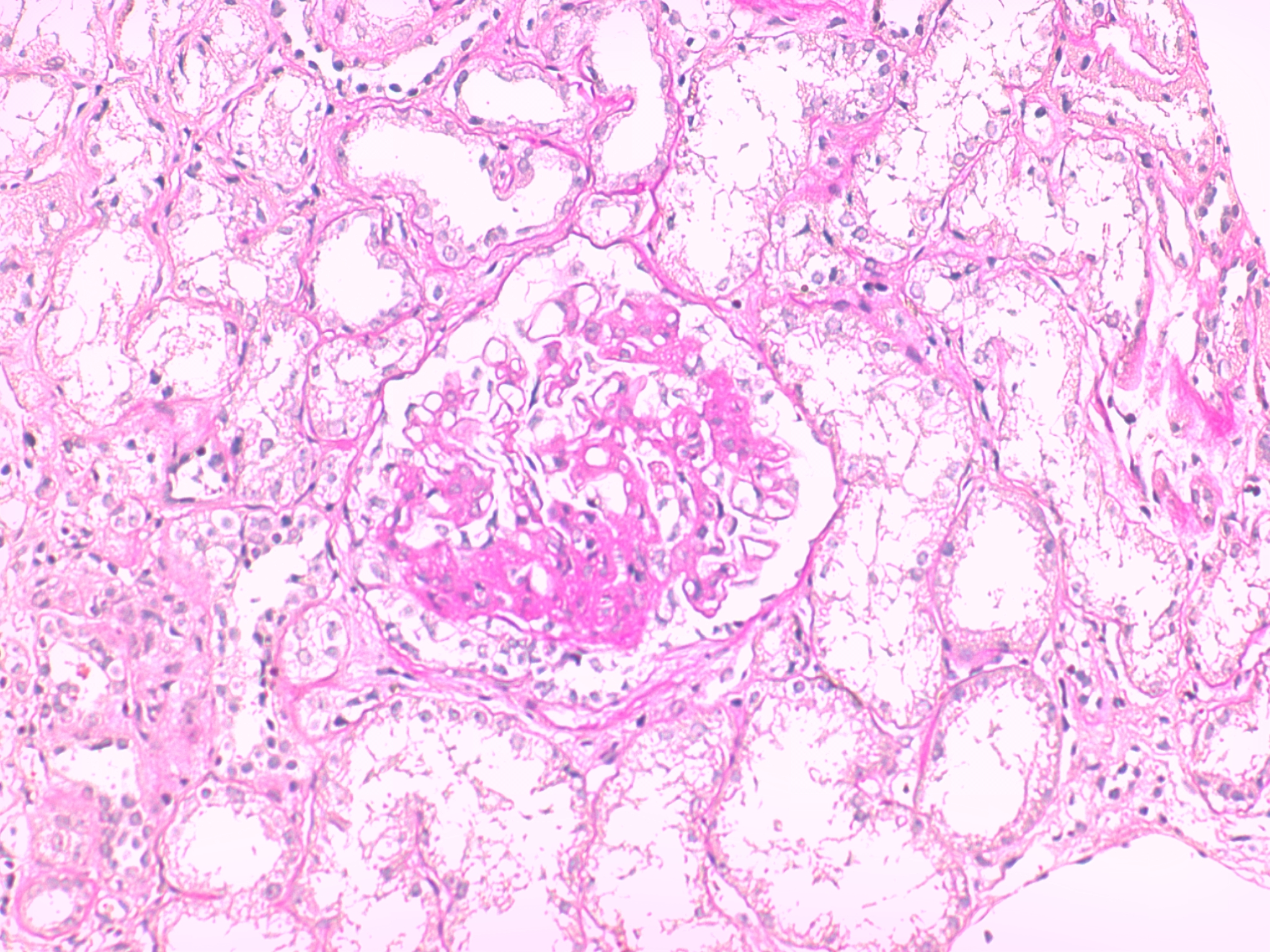
PAS com esclerose e paredes espessadas.
Figura IV
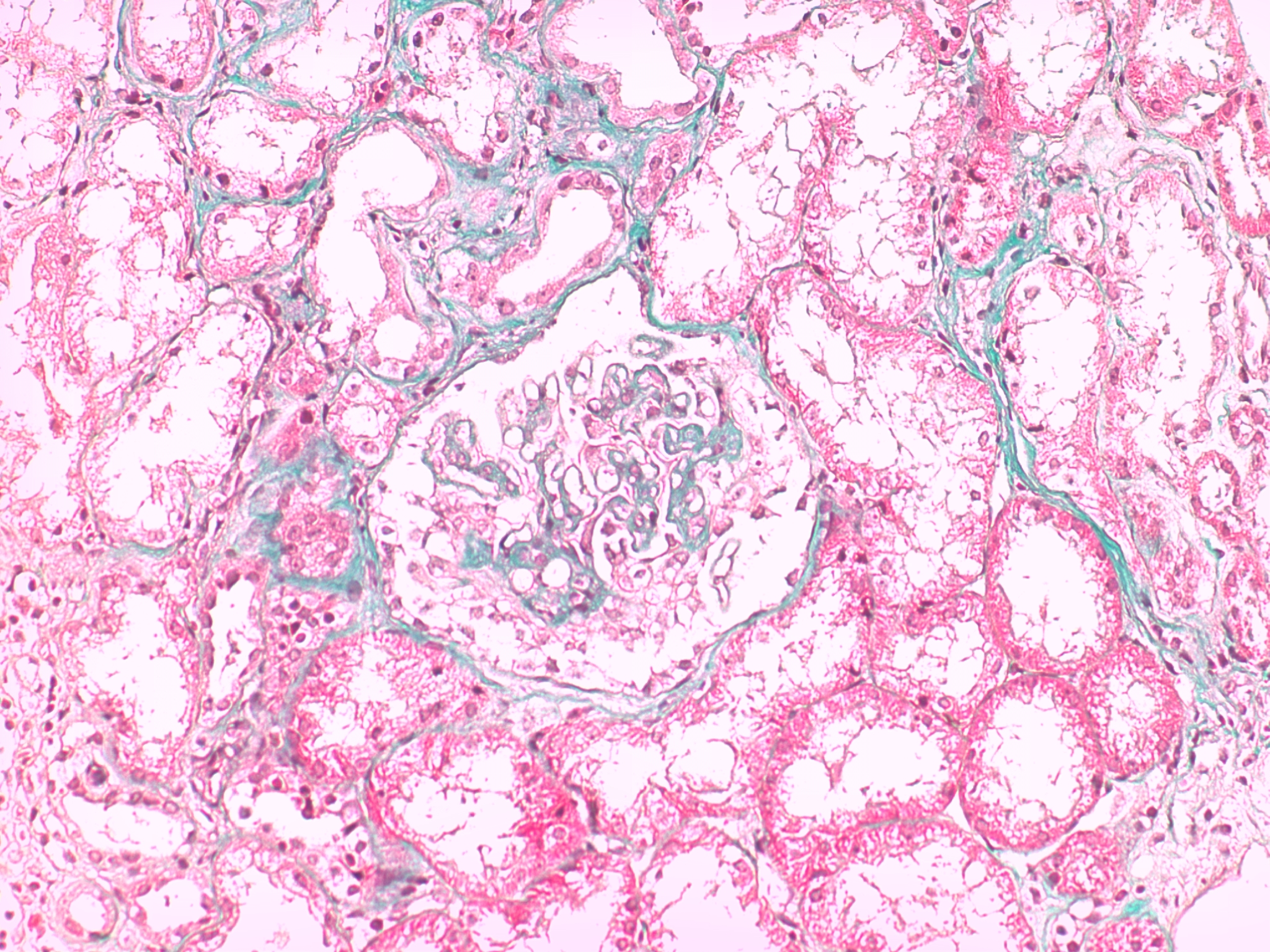
Tricrómio com paredes espessadas e hipertrofia de podócitos.
Figura V
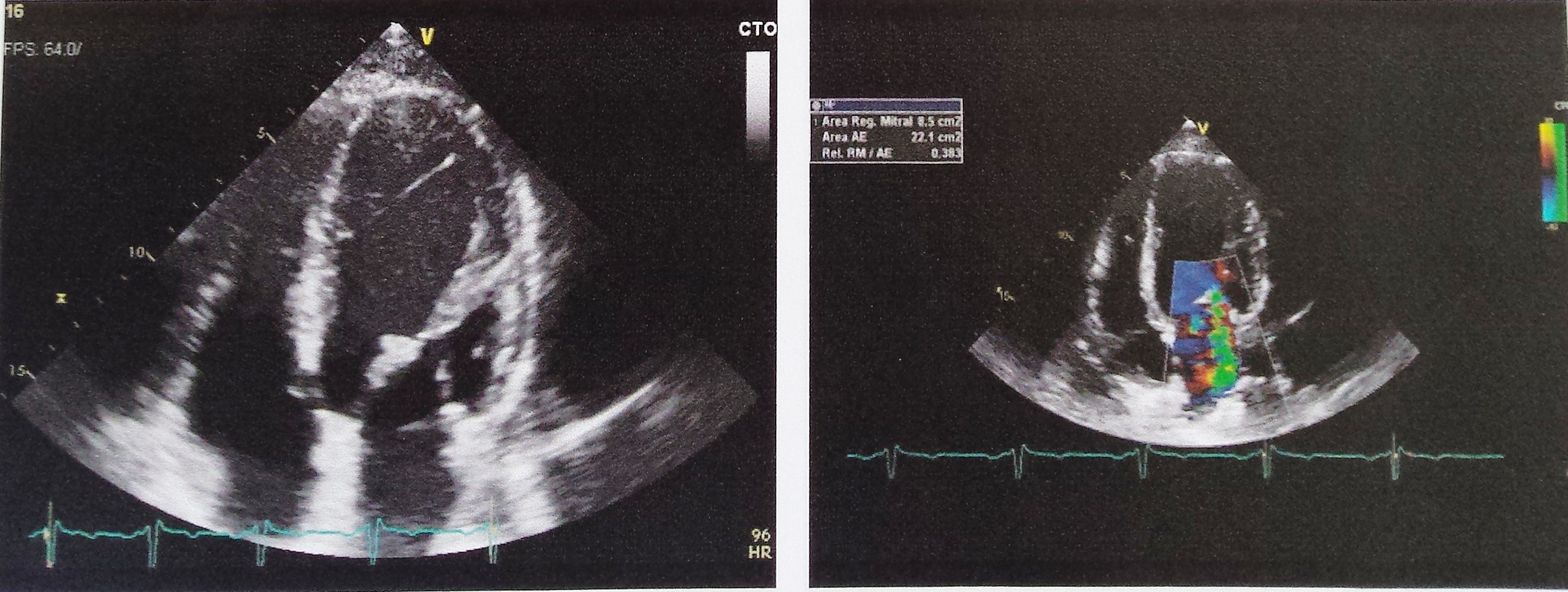
Ecocardiograma transesofágico com vegetações ao nível da válvula mitral compatíveis com endocardite de Libman-Sacks.
BIBLIOGRAFIA
1. Campos MM. Síndrome de hipoprotrombinémia -Anticogulante Lúpico. Acta Med Port. 2011; 24(S3): 611-16.
2. Devreese KMJ. Antiphospholipid antibodies: Evaluation of the thrombotic risk. Thrombosis Research. 2012; 130:S3740.
3. Hughes GRV. Thrombosis, abortion, cerebral disease and the lupus anticoagulant. BMJ. 1983; 287: 1088-9.
4. Nayer A, Ortega LM. Catastrophic antiphospholipid syndrome: a clinical review. J Nephropathol. 2014; 3(1): 9-17.
5. Gómez-Puerta JA, Cervera R. Diagnosis and classification of the antiphospholipid syndrome. J Autoimmun. 2014;48-49:20-5.
6. Abreu MM et al. The relevance of non-criteria clinical manifestations of antiphospholipid syndrome: 14th International Congress on Antiphospholipid on Antiphospholipid Antibodies Task Force Report on Antiphospholipid Syndrome Clinical Features, Autoimmun Rev. 2015; 14(5):401-14.
7. Ferreira E, Bettencourt PM, Moura LM. Valvular lesions in patients with systemic lupus erythematosus and antiphospholipid syndrome: An old disease but a persistent challenge. Rev Port Cardiol. 2012;31(4):295-9.
8. Sciascia S, Cuadrado MJ, Khamashta M, Roccatello D. Renal involvement in antiphospholipid syndrome. Nat Rev Nephrol. 2014; 10:279-84.
9. Mubarak M, Nasri H. What nephrolopatgologists need to know about antiphospholipid syndrome-associated nephropathy: is time for formulating a classification for renal morphologic lesions? J Nephropathol. 2014; (3):4-8.
10. Bomback AS, Markowitz Gs. Lupus Podocytopathy: A Distinct Entity. Clin J Am Soc Nephrol. 2016; 11(4):547-8.
11. Erkan D, Aguiar CL, Andrade D, Hannah Cohen, Maria J et al. 14th International Congress on Antiphospholipid Antibodies Task Force Report on Antiphospholipid Syndrome Treatment Trends. Autoimmun Rev. 2014; 12: 68596.
12. Galli M. Treatment of the antiphospholipid syndrome. Autoimmun Highlights. 2014; 5:17.