

INTRODUÇÃO:
As doenças cerebrovasculares de pequenos vasos correspondem a cerca de um quarto de todos os Acidentes Cerebrovasculares Isquémicos (AVC) e representam uma importante causa de deterioração cognitiva da população adulta, contribuindo para o aumento da incidência de demência na população geral. Na maioria dos casos, tanto os AVC como a Demência estão em associação directa com factores de risco cardiovascular, contudo, num pequeno subgrupo de indivíduos, estas doenças têm subjacente uma causa genética. A Arteriopatia Cerebral Autossómica Dominante com Enfartes Subcorticais e Leucoencefalopatia, conhecida pelo acrónimo de CADASIL é a arteriopatia cerebral hereditária de pequenos vasos mais comum1. É uma doença rara, caracterizada por acidentes cerebrovasculares, demência, depressão e enxaqueca com aura, afectando geralmente o adulto jovem (3º e 5º décadas de vida) e condicionando uma redução da esperança média de vida, geralmente de 65 anos no homem e 74 na mulher1. A CADASIL é o resultado de alterações microvasculares das células do músculo liso vascular, decorrentes da mutação autossómica dominante do gene NOTCH3 localizado no cromossoma 19, ocasionando oclusão das artérias cerebrais de pequeno calibre e promovendo o desenvolvimento de áreas de desmielinização2,3. Como qualquer doença genética, não existe nenhum tratamento específico para a CADASIL. O tratamento assenta na modificação de factores de risco cardiovascular associados e à terapêutica sintomática.
CASO CLINICO:
Mulher de 48 anos, caucasiana, com antecedentes de hipertensão arterial controlada de 7 anos de evolução e sem antecedentes familiares relevantes nomeadamente AVC, enxaquecas recorrentes ou demência em idades jovens, foi saudável até aos 20 anos. Nessa altura, iniciou quadro de cefaleias frontais opressivas contínuas e diárias de horas de duração, sem outros sintomas associados e que melhoraram com terapêutica com AINES. Aos 24 anos apresentou episódio de paralisia facial periférica pós parto, submetida a corticoterapia, da qual manteve sequelas (Lagoftalmia esquerda). Aos 43 anos reiniciou quadro de cefaleias, nessa altura, parieto-temporais esquerdas, lancinantes, de segundos de duração não associadas a outras manifestações neurológicas e com intensidade e duração crescentes ao longo de um mês, que a motivaram a procurar o médico assistente. Do estudo analítico e imagiológico com Tomografia Axial Computorizada Crânio-Encefálica (TAC-CE) realizado não se objectivou qualquer achado patológico. As cefaleias permaneceram com estas características por mais alguns meses, cedendo espontaneamente. Permaneceu assintomática por mais de 2 anos. Aos 47 anos recorreu ao médico assistente, 24h após apresentar novo episódio de cefaleia parieto-temporal esquerda, precedida de aura visual, ao qual se sucedeu episódio de perda súbita de força no membro inferior esquerdo, com recuperação ao fim de 2-3 minutos. Realizou TAC-CE que descreveu hipodensidades da substancia branca bilaterais e lesão cística parietal esquerda. Na sequencia desses achados, foi complementado o estudo com Ressonância Nuclear Magnética encefálica (RNM-E) que mostrou áreas hiperintensas da substancia branca periventricular bilateral, com predomínio à esquerda na sequencia T2. Após estes achados não manteve seguimento médico, aguardando consulta de Neurologia. Um ano depois, recorreu ao Serviço de Urgência na sequência de episódio de perda brusca da sensibilidade na face lateral do pescoço à esquerda e coxa esquerda, com recuperação em escassos minutos e sem descrição de cefaleia associada. À admissão encontrava-se hemodinamicamente estável, apirética, normoglicémica. À auscultação cardíaca rítmica com S1 e S2 presentes e sem sopros audíveis, não se auscultaram sopros carotídeos. Os pulsos periféricos eram amplos e simétricos. No exame neurológico sem défices motores, sensitivos ou de pares craneanos, para além da dificuldade em fechar o olho esquerdo. Os reflexos osteotendinosos eram vivos e simétricos nos dois membros inferiores e o reflexo cutâneo plantar era indiferente bilateralmente. Não apresentava síndrome cerebelosa. As análises encontravam-se dentro dos parâmetros da normalidade e a TAC-CE não veio revelar achados de novo para além da leucoencefalopatia anteriormente descrita. Face à clinica, antecedentes e anteriores achados imagiológicos foi proposto internamento para estudo. Analiticamente não apresentava alterações de hemograma, ionograma e bioquímica geral, com estudo de trombofílias negativa para Anticorpos anticardiolipina e anti-β2 Glicoproteína I, resistência de proteína C activada, pesquisa da mutação do gene de protrombina, Factor V de Leyden e Anticorpos antinucleares. Serologias virais foram negativas (HIV, VHC, VHC, CMV, EBV, Borrelia, VDRL). Repetiu RNM-E que descreveu Zonas de hipersinal difusa da substancia branca adjacente aos ventrículos laterais nos seus pisos infra e supra-tentoriais nas sequências de ponderação T2, com zona de hiposinal em T1 com localização adjacente ao ventrículo lateral esquerdo(Fig.1-2). A angio-RNM não detectou outras anomalias vasculares. Realizou também Electroencefalograma que não mostrou focalidade neurológica. No estudo, foi realizada punção lombar com análise de Liquido cefalorraquidiano (LCR), sem leucócitos e escassos eritrócitos, bioquimicamente normal, sem evidência de pico monoclonal de tipo IgG.
Foi solicitada também avaliação neuropsicologica, que revelou que a doente apresentava algumas dificuldades de atenção, de conceptualização e uma apraxia construtiva, levantando a suspeita de Síndrome frontal.
A associação de cefaleias, clinica de AIT, a existência de um síndrome frontal e de anomalias da substancia branca periventricular em exames imagiológicos fez-nos levantar a hipótese de CADASIL. Foi então solicitado estudo genético com sequenciação do gene NOTCH 3 (cromossoma 19), que revelou mutação do exão 11 desse gene. Iniciou terapêutica com perindopril 4 mg e AAS 100 mg/id., para optimização de controlo de factores de risco cardiovasculares. No controlo evolutivo anual, manteve queixas ocasionais de cefaleias esquerdas e agravamento das alterações da substancia branca cerebral na RNM encefálica com a atingimento simétrico da protuberância, talamos e gânglios da base.
DISCUSSÃO:
Este caso documenta uma causa rara de AVC no adulto jovem. O quadro neurológico inicial caracterizou-se por cefaleias frontais recorrentes e opressivas que evoluíram para cefaleias temporais com aura visual com características de enxaqueca, a que se associaram sintomas neurológicos focais transitórios como hipostesia ou perda da força segmentar transitória, sugestivos de AIT. Esta situação, podia levantar a hipótese de Esclerose Múltipla (EM), sobretudo pelas manifestações clinicas de focalidade neurológica em adulto jovem. No entanto, a diferente localização das placas desmielinizantes da esclerose múltipla na RNM-E (tipicamente ovóides e perpendiculares aos ventrículos e corpo caloso, com possibilidade de afectação cortical) associadas á presença de bandas oligoclonais na análise de LCR e potenciais evocados sugestivos, que não se confirmaram nesta doente bem como a evolução autolimitada, tornaram este diagnóstico pouco provável. A existência de clinica sugestiva de AIT a acrescentar á idade jovem da doente, obrigou a excluir trombofilias associadas. Outra entidade que obrigou o diagnóstico diferencial foi a encefalopatia subcortical de Binswanger. Esta forma de demência vascular, á semelhança da CADASIL, causa desmielinização e rarefacção da substância branca periventricular e enfartes lacunares. No entanto, distingue-se desta por afectar indivíduos com idade superior a 60 anos, sem que exista enxaqueca ou antecedentes familiares associados. Na Hidrocefalia normotensiva a associação de demência, alterações da marcha, incontinência de esfíncteres com os achados imagiológicos de alteração de substância branca periventricular, podiam confundir o diagnóstico diferencial de uma CADASIL em estadio avançado. Apesar de ponderadas outras hipóteses diagnósticas nomeadamente, doença infecciosa ou inflamatória, elas foram desde logo infirmadas pelas serologias para os principais vírus e culturas de LCR negativas, bem como pela negatividade dos estudos de autoimunidade. A cronicidade das lesões da substância branca periventricular, distribuídas de forma simétrica e bilateral, associadas a enxaqueca com aura e evidência de deterioração cognitiva4 em adulto jovem, veio levantar a possibilidade de CADASIL. Apesar da inexistência de contexto familiar sugestivo, existia uma forte suspeita de Arteriopatia Subcortical de causa genética. A determinação da sequenciação genética do gene NOTCH3, veio confirmar a mutação do exon 11.
A CADASIL é a doença cerebrovascular de causa genética mais comun5,6. Causada pela mutação monogénica, autossómica dominante6, do gene NOTCH 3 localizado no braço curto do cromossoma 195, que condiciona alterações da microestrutura vascular dos pequenos vasos cerebrais, manifestando-se clinicamente como AVC, AIT recorrentes, enxaqueca com aura, depressão e demência4. A prevalência da doença é ainda desconhecida, não só pela escassez de estudos mas também pela sua grande variabilidade fenotípica nos diferentes grupos étnicos7, inclusive entre indivíduos da mesma família8. Num estudo Escocês estimou-se que a prevalência da CADASIL nesse país foi de cerca de 1,98 casos por cada 100.000 adultos e que a prevalência da mutação foi de 4.14 por cada 100.000 adultos7. Antes do desenvolvimento de testes genéticos, a CADASIL foi frequentemente confundida com a Encefalopatia Subcortical de Binswanger, Hidrocefalia normotensiva9, Esclerose Múltipla, Doença de Alzheimer e outras doenças neurodegenerativas7. É actualmente uma importante área do investimento da Neurogenética. Apesar de mais de 150 mutações já terem sido descritas7,10, relacionadas com o gene NOTCH3, todas elas interferem na codificação do receptor de proteína transmembrana de Epidermal Growth Factor like-Repeat domain (EGFR) especifico da vasculatura das pequenas artérias cerebrais perfurantes11, condicionando alterações degenerativas e fibróticas das células do músculo liso vascular1,11 (CMLV), associada ao depósito de material osmiofílico granular (GOM) na parede do vaso1,7,8,12. Estas alterações condicionam redução do fluxo sanguíneo vascular e interferindo com os mecanismos de autorregulação cerebral8(12),13, originando, por conseguinte, isquémia cerebral. O diagnóstico clinico baseia-se no início dos sintomas compatíveis com enfartes lacunares entre 30-50 anos, em indivíduos sem factores de risco cardiovascular, e frequentemente com associação familiar7. O diagnóstico é feito com base nas características especificas da RNM-E (Hiperintensidades dos lobos temporais e da substância branca periventricular, simétrica e bilateral, enfartes lacunares no tálamo, centro semioval, gânglios da base e protuberância e microhemorragias cerebrais)7,14 associado á confirmação histopatológica do GOM obtidos por biopsia (nervo periférico, musculo ou pele) bem como estudo genético7. O tratamento não se encontra bem definido8,10. Vários estudos documentam que o estrito controlo de factores de risco cardiovasculares pode atrasar o inicio dos sintomas incapacitantes da doença (depressão, demência, dependência)8. Neste sentido o Acido acetilsalicílico e controlo estrito de tensão arterial mostraram ser benéficos. Os anticoagulantes, pelo contrário, não estão recomendados dado o aumento do risco hemorrágico. As crises de enxaqueca podem tratar-se com AINES ou analgésicos convencionais, uma vez que os ergóticos e triptanos, pelo seu efeito vasoconstritor podem ser deletérios na CADASIL. Como medida profiláctica da enxaqueca pode optar-se por Beta-bloqueantes, antagonistas dos canais de cálcio ou antiepilépticos. A demência, como manifestação integrante desta síndrome, está relacionada com os eventos isquémicos recorrentes. Os inibidores da acetilcolinesterase mostraram ligeiro benefício em doentes com deterioração cognitiva9,10.
Agradecimentos: Os autores agradecem ao Dr. Pedro Rosado, Neurologista do Centro Hospitalar Cova da Beira, pelos conselhos e ajuda prestada na realização deste trabalho.
Figura I
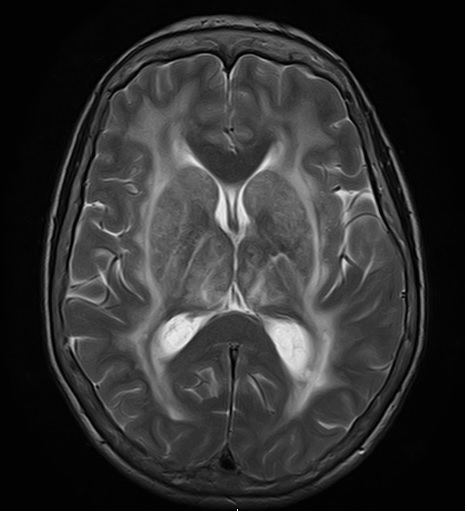
Corte axial de RNM craneoencefalica em ponderação T2 com areas hiperintesnsas da susbstância branca periventricular bilateral sugestivas de CADASIL
Figura II
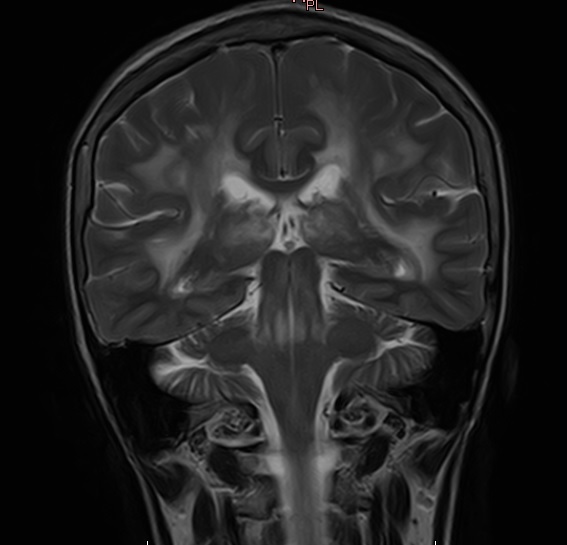
Corte coronal de RNM craneoencefalica em ponderação T2 com areas hiperintensas da susbstância branca periventricular bilateral sugestivas de CADASIL
BIBLIOGRAFIA
1.Andre C, CADASIL: Pathogenesis, clinical and radiological findings and treatment. Arq Neuropsiquiatr.2010 Apr;68(2):287-99.
2.Opherk C, Peters N, Herzog J, Luedtke R, Dickgans M. Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain, 2004 Nov; 127 (pt11):2533-9.
3.Caplan R, Louis. Lacunar Infarction and Small Vessel Disease: Pathology and Pathophysiology. Journal of Stroke 2015;17(1):2-6.
4.Henao-Arboleda E, Aguirre-Acevedo DC, Pacheco D. Seguimiento de las características cognitivas en una población con enfermedad cerebrovascular hereditaria (CADASIL) en Colombia; REV NEUROL 2007; 45 (12): 729-733.
5.Chol Choi J, Song S ; Lee JS; Kang SY and Kang J-H. Headache among CADASIL patients with R544C mutation: Prevalence, characteristics, and associations; Cephalalgia 2014, Vol 34(1) 2228.
6.Yamamoto Y, Craggs LJ, Watanabe A, Booth T, Attems J, Low RW. Brain microvascular accumulation and distribution of the NOTCH3 ectodomain and granular osmiophilic material in CADASIL. J Neuropathol Exp Neurol 2013;72(5):416-431.
7.Stojanov D, Aracki-Trenkic A, Vojinovic S; Ljubisavljevic S, Benedeto-Stojanov D; Tasic A, Vujnovic S. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). Bosn J Basic Med Sci. 2015;15(1):1-8.
8.Ciolli L, Pescini F, Salvadori E, Del Bene A, et all. Influence of vascular risk factors and neuropsychological profile on functional performances in CADASIL: results from the Microvascular Leukoencephalopathy Study (MILES). European Journal of Neurology 2014, 21: 6571.
9.Vásquez do Campo R, Morales-Vidal S, Randolph C, Chadwick L. CADASIL: descripción de una serie de 11 casos clínicos. Rev Neurol 2011,52 (4): 202-210.
10.Rutten-Jacobs, Loes CA , Traylor M ; Adib-Samii P, Thijs V, Sudlow C, et all. Common NOTCH3 Variants and Cerebral Small-Vessel Disease. Stroke. 2015;46:1482-1487
11.Zhu Y, Wang J, Wu Y, Wang G, Hu Bai. Two novel mutations in NOTCH3 gene causes cerebral autosomal dominant arteriopathy with subcritical infarct and leucoencephalopathy in two Chinese families. Int J Clin Exp Pathol 2015;8(2):1321-1327.
12.Arcos-Burgos M, Restrepo T, Rivera D, Palacio LG, et all. Demencia vascular hereditária tipo CADASIL en Colombia. III. Análisis de ligamiento a Notch3. REV NEUROL 2001;32(8):701-704.
13.Noh S-M; Chung SJ; Kim K-K; Kang D-W. Emotional Disturbance in CADASIL: Its Impact on quality of Life and Caregiver Burden. Cerebrovasc Dis 2014;37:188-194.
14.Yamamoto Y, Ihara M, Tham C, Low RW, Slade JY, Moss T, et all. Neuropathological correlates of temporal white matter hyperintensities in CADASIL. Stroke 2009;40(6):2004-2011.