

HEMOFILIA A ADQUIRIDA:RELATO DE CASO
Introdução
A hemofilia A adquirida (HAA) é uma coagulopatia adquirida auto-imune 1. Pode ocorrer em indivíduos de qualquer idade sem história prévia de hemorragia2 (ver Fig. 1). Está associada a uma heterogeneidade de quadros clínicos, nomeadamente doenças auto-imunes3,4, período pós-parto (tipicamente nos 2-3 meses posteriores)5, neoplasias e fármacos. Em cerca de 50% dos casos não se consegue determinar fator etiológico predisponente2.
A sua incidência varia entre 1-1,5 casos/milhão de habitantes/ano 6. Tem uma distribuição bifásica em relação à idade, com um pico entre os 20 e os 40 anos predominante em mulheres, devido aos casos de HAA no período pós-parto e outro pico em adultos a partir dos 65 anos.
Muito frequentemente os doentes apresentam-se com hematomas extensos, equimoses, hemorragias das mucosas (epistaxes, hemorragia gastrointestinal, hematúria)2, sendo raro envolvimento articular.
O diagnóstico baseia-se num estudo de coagulação anormal, com um tempo de tromboplastina parcial ativado (aPTT) geralmente prolongado isoladamente, que não corrige com o teste de mistura (ver Fig. 2). Um aPTT prolongado com um tempo de protrombina (TP) normal indica que existe um défice de um dos fatores da via intrínseca da coagulação (F VIII, IX,XI, XII) ou presença de anticorpo antifosfolipídico, não indicando qual o fator diminuído 7. No teste de mistura (plasma do doente com plasma normal) incubado a 37° durante 1-2 horas, na presença de um inibidor o aPTT não corrige, posteriormente adiciona-se uma fonte de fosfolípidos no plasma misturado, se houver correção do aPTT sugere a presença de anticorpo antifosfolipídico 8. Existe ainda o teste de Bethesda que estabelece simultaneamente o diagnóstico da presença de inibidor de FVIII e quantifica o título de anticorpo 9. Neste teste são efetuadas incubações a 37° durante 2 horas de plasma normal com plasma do doente, definindo-se como uma unidade de inibidor a quantidade de inibidor que inativa metade de FVIII existente na mistura9.
O tratamento da HAA tem quatro objetivos principais: prevenção da hemorragia, controlo da hemorragia, erradicação do inibidor e investigação e tratamento da eventual causa subjacente.
O tempo mediano para remissão é em média 6 semanas utilizando corticoterapia, com ou sem uso de ciclofosfamida 11,12. Deve ser continuado pelo período de 2 meses, sendo o doente monitorizado com aPTT pelo menos 1 ano após a terapêutica13.A mortalidade é elevada (cerca de 22%), podendo estar relacionada com a hemorragia ou efeitos do próprio imunossupressor (exemplo sépsis) 13,14.
Relato de caso
H.D.R.A, 72 anos, sexo masculino, caucasiano, internado no Serviço de Medicina Interna IA do Hospital de Egas Moniz, por quadro de discrasia hemorrágica grave.
O doente recorreu ao Serviço de Urgência por quadro de hemorragia espontânea de úlcera arterial da face anterior do membro inferior esquerdo com 1 dia de evolução e de agravamento progressivo.
O doente apresentava múltiplas comorbilidades: diabetes mellitus tipo 2, hipertensão arterial essencial, doença renal crónica estadio 3a, fibrilhação auricular, insuficiência cardíaca de etiologia mista (isquémica, valvular), estenose aórtica moderada, anemia da doença crónica, doença arterial periférica, hiperuricemia e dislipidemia. A referir dois internamentos prolongados nos últimos 3 meses no contexto de infeções respiratórias, sem agente etiológico identificado. O último antibiótico foi Piperacilina/Tazobactam cerca de 1 mês antes do internamento descrito neste relato de caso. Estava polimedicado com amlodipina/valsartan, bisoprolol, furosemida, insulina, linagliptina, sinvastatina, alopurinol, pentoxifilina e buprenorfina.
Ao exame objetivo destacava-se hematoma sangrante da face anterior da perna esquerda, sem hemorragia pulsátil, 7*12 cm.
Analiticamente apresentava: hemoglobina 8.1 g/dl (valor de 10 g/dl nos 2 meses anteriores), VGM 78.1 fL, HGM 24.7 pg, ferritina 879 µg/ml, aPTT 91 segundos (controlo = 29.5 segundos), TP normal, contagem plaquetária normal, anticoagulante lúpico negativo, F VIII inferior a 1% e posteriormente a presença de inibidor para o F.VIII (558 Unidades Bethesda).
Perante o quadro clinico foi assumida hemofilia A adquirida e iniciou terapêutica com corticoterapia (prednisolona 1 mg/kg/dia). Face à necessidade de controlo da hemorragia fez terapêutica com CPA.
Durante o internamento não se verificaram novos episódios de hemorragia aguda que justificassem necessidade de administração de complexo protrombínico ativado (ver Fig. 3). Verificou-se descida progressiva do aPTT, à data da alta de 54.8 segundos e subida do F VIII para 3%.
Para investigação adicional de coagulopatia adquirida, foi efetuado estudo analítico, nomeadamente de patologias de foro auto-imune sem alterações detectadas, nomeadamente ANA, ENAs e anticoagulante lúpico normais. Efetuou ainda eletroforese de proteínas interpretada como normal, assim como os doseamentos de imunoglobulinas. Os marcadores víricos foram negativos (HIV, HBV, HCV).
A destacar que o doente havia sido submetido a exames complementares recentes com função tiroideia, antigénio prostático especifico (PSA), tomografia computorizada toraco-abdómino-pélvico e colonoscopia total sem alterações a destacar.
Teve alta sem evidência de discrasia hemorrágica, indicação para manutenção de prednisolona 1 mg/kg e reavaliação em consulta externa de Medicina Interna, Imunohemoterapia e Cirurgia Vascular.
Discussão
A HAA geralmente cursa com inicio súbito, com hemorragias espontâneas em mais de 70% dos casos. A sua etiologia está relacionada com a produção de autoanticorpos anti-F VIII 1, são geralmente da classe IgG. A apresentação clínica está relacionada com uma sintomatologia hemorrágica geralmente grave, que necessita de diagnóstico e tratamento emergentes.
O tratamento imunossupressor resulta na remissão da doença em 60-80% dos doentes 1. A severidade da hemorragia à apresentação não é preditivo de futuras hemorragias e os doentes permanecem em risco de hemorragia até à irradicação do inibidor 7,10.
Quanto à prevenção da hemorragia devem ser evitados procedimentos invasivos desnecessários (até à eliminação do inibidor) bem como terapêutica antiagregante plaquetária, anticoagulante e antiinflamatórios não esteróides.
No que diz respeito ao controlo da hemorragia, é a gravidade desta que deve condicionar a opção do tratamento hemostático. Em situações graves deve ser ponderada a terapêutica de by-pass recorrendo ao uso de FVIIa (Fator VII ativado) e CPA (Complexo Protrombinico Ativado - FEIBA). Se estes recursos não estiverem disponíveis pode ser equacionado o uso de concentrados de F VIII em alta dose e até eventualmente DDAVP (desmopressina)7,10. Deve ter-se em atenção o volume de perdas sanguíneas e sua eventual reposição adequada.
Quanto à erradicação do inibidor do F VIII é necessário o recurso a terapêutica imunossupressora. Está recomendada que esta seja iniciada assim que esteja estabelecido o diagnóstico de HAA. Como tratamento de primeira linha é recomendado a administração de prednisolona numa dose de 1 mg/kg peso/dia13 associado ou não a ciclofosfamida numa dose de 1,5-2 mg/kg peso/dia. Não existe actualmente nenhum estudo randomizado que demonstre superioridade de um regime imunossupressor especifico. Como tratamento imunossupressor de 2ª linha há que considerar o Rituximab (375 mg/m2 superfície corporal). Pode utilizar-se em monoterapia ou em associação com ciclofosfamida ou associação com ciclofosfamida mais prednisolona. Como tratamentos alternativos há a considerar a azotioprina, vincristina, micofenolato e ciclosporina 7,10.
No caso descrito não se conseguiu estabelecer relação causal para o desenvolvimento de autoanticorpos, exceto terapêutica prévia recente com múltiplos antibióticos. Admitindo-se um fármaco como fator causal de HAA após a sua suspensão o título de anticorpos deveria baixar, no entanto faltam estudos que indiquem qual o tempo mediano necessário para essa diminuição. O último ciclo de antibioterapia do doente em questão foi cerca de 1 mês antes do início deste quadro.
Para o diagnóstico foi importante a apresentação clínica, aliada aos achados laboratoriais de aumento isolado de aPTT e identificação posterior de inibidor de F VIII. É fulcral a valorização precoce, quer pelo impato direto no prognóstico imediato, como em termos de mortalidade.
As opções terapêuticas são variadas, dependente também do título de inibidor existente. As opções terapêuticas disponíveis têm um perfil de validação débil, tendo em conta o caráter raro da doença, que torna difícil a realização de estudos comparativos. Apesar disso a utilização de prednisolona está bem estabelecida e foi esta a opção de tratamento.
Chama-se a atenção para a presença de fatores de mau prognóstico neste doente, nomeadamente comorbilidades graves, polimedicação e risco infecioso elevado.
Conclusão
Sendo esta entidade clinica diagnosticada tardiamente há que ter presente a importância de um diagnóstico atempado. Deve ser equacionado em doentes com episódio de discrasia hemorrágica, sem história prévia de hemorragia, aPTT prolongado e TP normal.
A identificação desta situação deve implicar sempre a sua investigação etiológica.
Seria importante, por tudo o que foi previamente descrito, a criação de um registo de casos a nível nacional.
Agradecimentos
Os autores agradecem ao Serviço de Imunohemoterapia do Hospital de Egas Moniz toda a colaboração.
Figura I
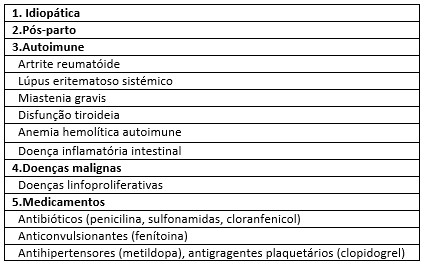
Condições associadas a HA
Figura II
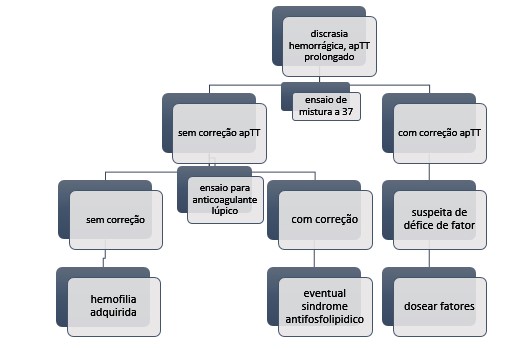
Algoritmo de diagnóstico de HAA
Figura III
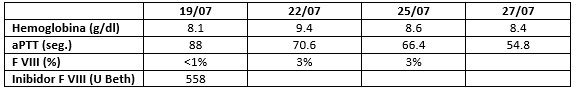
Evolução laboratorial durante o internamento
BIBLIOGRAFIA
Bibliografia
1. Tiede, A. et al. Prognostic factors for remission of and survival in acquired hemophilic A (AHA). GTH-AH 01/2010 study. Blood Journal, 2015. 125(7)
2. Franchini M, Mannuci PM. Acquired haemophilia A: a 2013 update. Thromb Haemost.2013; 110 (6): 1114-1120.
3. Green D, Lechner K. A survey of 2015, non hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981; 45-200.
4. Green D Schuette PT, Wallace Wtt. Factor VIII autobodies in rheumatoid arthritis. BM J Rheumatol 1987; 26-381.
5. Francini M. Postpartum acquired factor VIII inhibitors. Am J Hematol 2006; 81-768.
6. Kessler CM, Knobl L. Acquired haemophilic: an overview for clinical pratice. Eur J Haematol. 2015 Dec. 95 Suppl 81: 36-44.
7. Chalmers, Elizabeth, et al. Diagnosis and management of acquired coagulation inhibitors: a guidline from UKHCDO. Bristish Journal of Haematology, 2013. 162, p.758-773.
8. Cabral Ana, Romão Teresa, et al. Hemofilia adquirida. Medicina Interna. Vol 5, Nº1, 1998.
9. Kitchen Steve, Mc Craw Augus, et al. Diagnosis of hemophilia and other bleeding disorders. 2º edition, 2010.
10. Kuhne Angela, Baudo Francesco, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haemotaologica. 2009; 94 (4).
11. Ludlom CA, Morrison AE, et al. Treatment of acquired haemophilia. Seminars in Hematology 1994; 2(2) 4:16-19.
12. http://www.fda.gov
13. Giangrande Paul. Acquired Hemophilia Revised Edition. Oxford Haemophilia & Thrombosis Centre Oxford. November 2012. No 38.
14. Collins P Bando F, Knoebi P, et al. Immunosupression for acquired hemofilia: results from the European Avquired Haemophilic Reistry (EACH2). Blood 2012; 120:47.