Introdução
A doença de Kikuchi-Fujimoto (DFK) também denominada Linfadenite Histiocítica Necrosante, é uma entidade rara, benigna, auto-limitada, de etiologia desconhecida, caracterizada pela presença de adenopatias cervicais na grande maioria dos casos associadas a sintomas constitucionais.2 Atinge preferencialmente o sexo feminino na segunda ou terceira década de vida e é mais frequente nos países asiáticos, embora tenha uma distribuição mundial. 1,2 A incidência real desta doença é estimada entre 0.5% a 5% de todas as adenopatias avaliadas histologicamente.1 É diagnosticada após a excisão e estudo histológico de um gânglio, devendo ser diferenciada de outras patologias cuja forma de apresentação possa ser semelhante, nomeadamente as linfoproliferativas, infecciosas e autoimunes.1,2,3 O tratamento da DKF é apenas sintomático.1
Caso Clínico
Doente do sexo feminino, com 38 anos de idade e raça parda, que recorreu ao Serviço de Urgência por apresentar desde as últimas duas semanas uma tumefacção cervical direita dolorosa e de crescimento progressivo (figura 1). Cerca de quatro dias antes de recorrer à Urgência, com aparecimento de febre elevada refractária à medicação antipirética e astenia. Sem outras queixas sintomáticas. Sem antecedentes pessoais e familiares relevantes. Sem história epidemiológica significativa. Ao exame físico na admissão, encontrava-se febril (39.3 ºC), corada, anictérica, sem exantemas e com bom estado geral. Apresentava adenopatias cervicais à direita, com atingimento da cadeia cervical posterior direita, dolorosas à palpação, de consistência dura, móveis e não aderentes aos planos profundos, tendo a maior cerca de 2 cm de diâmetro. Não eram palpáveis adenopatias axilares, supraclaviculares ou inguinais. O exame da orofaringe era normal, bem com os exames do tórax, abdómen, membros e coluna vertebral. O exame neurológico não revelou alterações. Analiticamente existia anemia com hemoglobina 10.5 g/dL e leucopenia (2.2x103/L) com neutropenia (46%). Sem alterações dos marcadores de citólise ou colestase hepática. TAC cérvico-torácica e abdominal a demonstrar a presença de múltiplas adenopatias laterocervicais direitas, medindo a maior 18 mm de eixo curto (figura 2). Baço globoso medindo 13 cm de diâmetro longitudinal. Fígado com contornos regulares, forma e dimensões preservadas. Ausência de derrame peritoneal, massas ou adenopatias abdominais. Foi internada no Serviço de Medicina Interna para esclarecimento etiológico mais célere da situação, tendo inicialmente sido levantadas as seguintes hipóteses diagnósticas como as mais prováveis: patologias infecciosas como o VIH (Vírus da Imunodeficiência Humana), Mononucleose infecciosa e Tuberculose, nomeadamente a Tuberculose gangIionar), patologias linfoproliferativas (Linfoma não Hodgkin/Doença de Hodgkin), doenças auto-imunes (Lúpus eritematoso sistémico). Do estudo inicialmente realizado, serologias de doenças infecciosas tais como VIH serotipos 1 e 2, Hepatite B e C, Epstein Barr (EBV) (IgM e IgG), Herpes I e II, Citomegalovírus (CMV) (IgM e IgG) e VDRL, negativas. A pesquisa de autoanticorpos (ANAs, anti-dsDNA e Anti-SM) também foi negativa. Hemoculturas com pesquisa de micobactérias negativas. Efectuada biópsia de medula óssea, cujo resultado descreveu quadro morfológico compatível com hiperplasia reactiva das três linhas hematopoiéticas. Sem outras alterações. Mieloculturas para micobactérias negativas. Realizada biópsia tru-cut de um gânglio cervical direito cujo exame histológico demonstrou parênquima de gânglio linfático com extensas áreas de necrose com detritos celulares e sem participação de polimorfonucleares neutrófilos, aspectos morfológicos sugestivos de Linfadenite Histiocítica Necrosante (Figura 3). Exame cultural para micobactérias de gânglio, negativo. Citometria de fluxo sem alterações compatíveis com neoplasias hematológicas. O resultado da biópsia, compatibilizado com a clínica que também poderia ser sugestiva de Doença de Kikuchi-Fujimoto, e através da exclusão de outras entidades diagnósticas potencialmente mais prováveis, confirmou assim o diagnóstico. A doente melhorou gradualmente ao longo do internamento, com controlo da dor através da terapêutica com anti-inflamatórios não esteróides. À data da alta, já se apresentava sem febre e com regressão parcial do tamanho das adenopatias. Quatro meses após o início dos sintomas, constatada regressão total do quadro sintomático.
Discussão
A doença de Kikuchi-Fujimoto (DKF), ou Linfadenite Histiocítica Necrosante, foi pela primeira vez descrita em 1972 no Japão, por Kikuchi e Fujimoto. É uma doença rara, sendo quatro vezes mais frequente no sexo feminino do que no sexo masculino, sobretudo na segunda e terceira décadas de vida.3 A febre surge como sintoma inicial em 30% a 50% dos doentes, embora o sintoma de apresentação mais frequente seja o aparecimento de linfadenopatias localizadas. Estas geralmente estão localizadas na cadeia jugulo-carotídea e triângulocervical posterior, tal como apresentado neste caso clínico, embora também possam surgir de forma generalizada e difusa. Os gânglios linfáticos têm habitualmente até 3 cm, embora possam alcançar os 5-6 cm de diâmetro.4 Sintomas como fadiga, artralgias, artrite, erupções cutâneas e hepatoesplenomegalia também podem ser encontrados. Apesar da etiologia permanecer actualmente desconhecida, vários autores defendem que a doença surge como uma reacção hiperimune induzida por diferentes estímulos antigénicos ou processos autoimunes nos quais a apoptose assume um papel importante. Alguns agentes infecciosos, tais com o EBV, CMV, Toxoplasmose, Brucella, Yersínia Enterocolítica, Herpes Simplex 1 e 2, Varicella Zoster, Herpes Vírus Humano tipo 6 e 8, Parvovírus B19, VIH, Parainfluenza e Hepatite B, têm sido propostos como possíveis estímulos antigénicos1. No entanto, a negatividade dos estudos serológicos e das pesquisas microbiológicas tornam esta hipótese controversa.3 A DKF tem geralmente início subagudo, desenvolvendo-se durante um período de 2 a 3 semanas. Os exames laboratoriais são muitas vezes normais. No entanto, podem surgir: anemia, neutropenia (em 50% dos casos), linfocitose, linfócitos atípicos (em 25% dos casos), trombocitopenia, elevação da velocidade de sedimentação, da proteína C reactiva e aumento da desidrogenase láctica e das enzimas hepáticas. No presente caso clínico, a doente apresentava apenas os dois primeiros. Os anticorpos antinuclear, anti RNP, anti- DNA e anticoagulante lúpico podem ser positivos.3 O diagnóstico da DKF é histológico e tem um espectro de apresentações histológicas, mediante o estadio da doença. Existem três estadios, precoce, intermédio e tardio, também referidos como proliferativo, necrosante e xantogranulomatoso. No estadio proliferativo observam-se agregados de células dendríticas plasmacitoides, células blásticas e histiócitos. A necrose é mínima ou ausente. No estadio necrosante, dependendo do grau de envolvimento, a arquitectura ganglionar está parcialmente preservada. A característica histológica mais evidente são áreas de necrose, que podem ser encontradas nas áreas interfoliculares ou paracorticais. No seio da necrose observam-se detritos celulares e células apoptóticas; não se identificam neutrófilos. A ausência de neutrófilos nesta fase é útil para o diagnóstico diferencial, sendo uma característica muito sugestiva de DKF. Neste caso clínico, a biopsia realizada ao gânglio apresentava aspectos histológicos compatíveis com este estadio da doença. No estadio xantogranulomatoso o tecido linfóide é maioritariamente substituído por histiócitos de citoplasma xantelasmizado e estruturas granulomatosas.5 Não existe tratamento eficaz para esta entidade clínica. Os sinais e sintomas geralmente desaparecem dentro de 4 meses, tal como se observou com a nossa doente. Os doentes com sintomas mais graves ou persistentes têm sido tratados com corticosteróides. Deve ser realizado follow-up a longo prazo devido à possibilidade de recorrência da doença (até 3% dos casos) e à possibilidade de desenvolvimento de Lúpus Eritematoso Sistémico.6,7
Conclusão
A doença de Kikuchi-Fujimoto, apesar de ser uma doença rara, benigna, e que habitualmente não é colocada como primeira hipótese diagnóstica, deve ser um diagnóstico diferencial a considerar em casos de febre e adenopatias cervicais em mulheres jovens, fundamentalmente por poder ser confundida com outras entidades patológicas graves, geralmente mais frequentes, mas com tratamentos agressivos.
Figura I
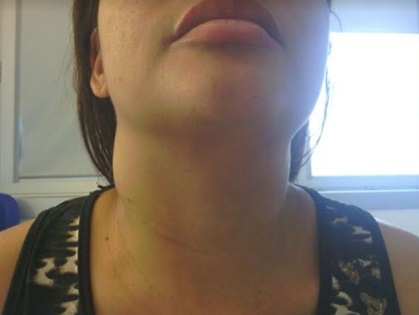
Conglomerado adenopático no triângulo posterior cervical direito.
Figura II
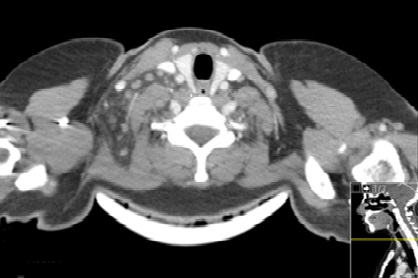
Imagem do TAC cérvico-torácico a demonstrar múltiplas adenopatias laterocervicais direitas com necrose central e fleimão circundante, sem abcedação.
Figura III
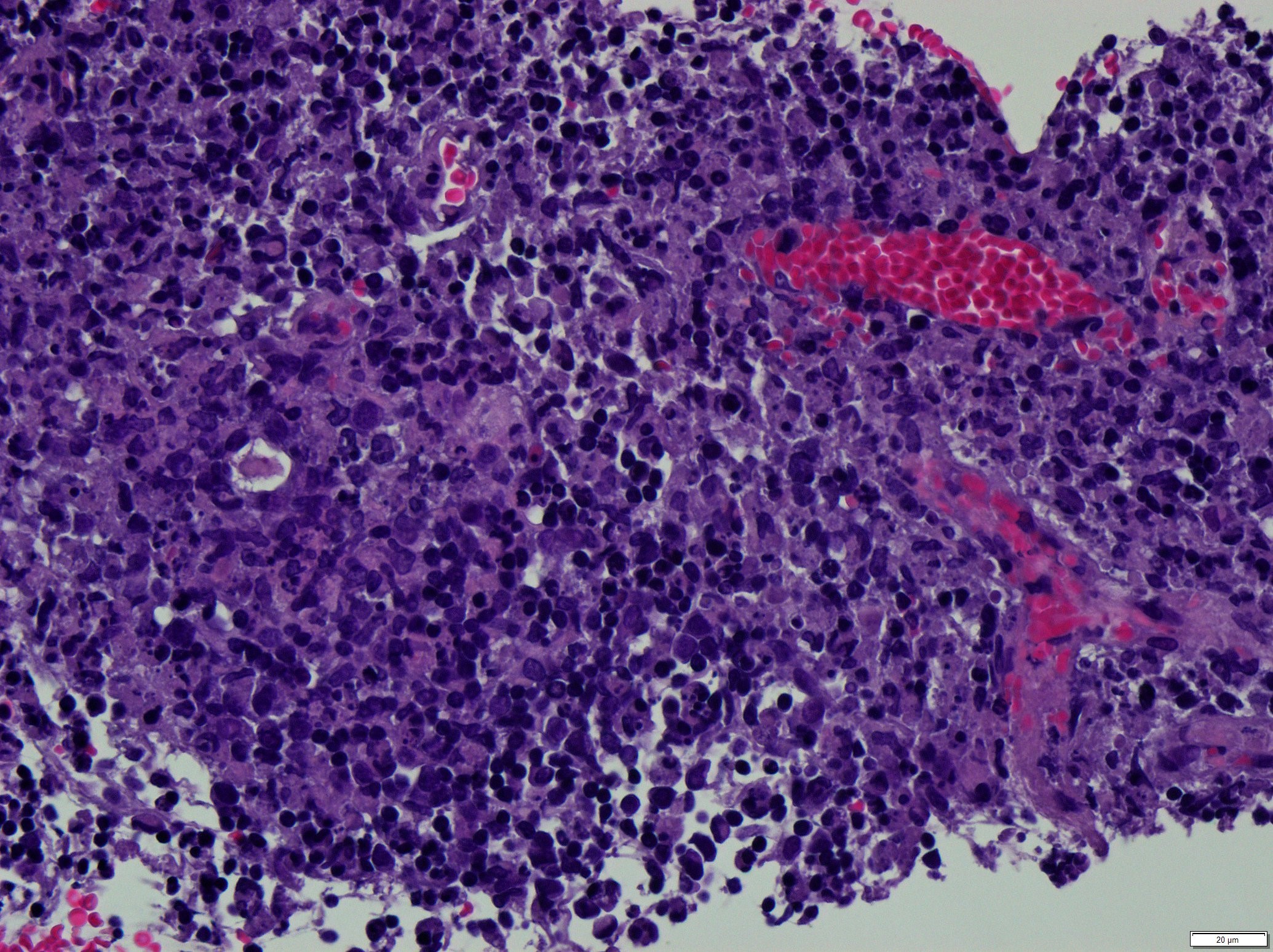
Área de necrose com detritos celulares. Não se identificam neutrófilos. (H&E, 100x).
BIBLIOGRAFIA
1. Antunes I, Botella A, Marques F, Araújo I, Abreu A, Cardiga R, Ceia F. Linfadenite Necrotizante (Doença de Kikuchi-Fujimoto), Acta Med Port 2011; 24, 681684.
2. Pina R, Fonseca I, Helena M. Doença de Kikuchi-Fujimoto: Uma causa pouco frequente de adenopatias cervicais, Med Interna 2004;11, 187190.
3. Gonçalves M, Silva G, Barros A, Freitas L, Caldeira P, Rodrigues R, Capelinha F, Freitas E, Fraga C, Camacho J, Ferreira A. Doença de Kikuchi-Fujimoto e Lúpus Eritematoso, Med Interna 2008; 112116.
4. Deaver D, Naghashpour, M, Sokol L. Kikuchi-fujimoto disease in the Unites States: Three case reports and review of the literature. Mediterranean Journal of Hematology and Infectious Diseases, 2014; 6(1). http://doi.org/10.4084/MJHID.2014.001
5. Orazi A, Weiss LM, Foucar K, Knowles DM. Knowles´ Neoplastic Hematopathology. 3rd ed. United States: Lippincot Williams & Wilkins; 2013. p.343-6.
6. Practice C, Schwalfenberg G. Case Report; Kikuchi-fujimoto disease. Canadian Family Physician, 2008; 54; 864866. http://doi.org/10.1007/s11999-008-0150-6
7. Dumas G, Prendki V, Haroche J, Amoura Z, Cacoub P, Galicier L, Fain O. Kikuchi-Fujimoto Disease. Medicine, 2014; 93(24), 372382. http://doi.org/10.1097/MD.0000000000000220

