

INTRODUÇÃO
A complexidade das doenças auto-imunes, caracterizadas por uma marcada heterogeneidade fenotípica e imunológica, tem o seu paradigma na sobreposição de perfis de auto-anticorpos e de manifestações clínicas de diferentes doenças num mesmo indivíduo1. A influência de factores genéticos e ambientais, bem como a partilha de mecanismos fisiopatológicos entre diferentes doenças, parecem estar na base das síndromes de sobreposição.
CASO CLÍNICO
Os autores descrevem o caso de uma mulher, com antecedentes de tabagismo activo, que, aos 22 anos de idade, foi referenciada à consulta de doenças Auto-Imunes por poliartralgias com dois anos de evolução. Referia acometimento dos punhos, 2ª e 3ª articulações metacarpo-falângicas bilateralmente, com rigidez matinal de duas horas, bem como edema discreto e assimétrico das articulações tibiotársicas. Descrevia alteração da coloração dos dedos das mãos, mais pronunciada à direita, compatível com fenómeno de Raynaud bifásico, fotossensibilidade e xerostomia. Negava sintomas constitucionais, rash malar, úlceras orais ou genitais, xeroftalmia, eventos trombóticos ou obstétricos, disfagia, lombalgia inflamatória, uveíte, diarreia sanguinolenta ou psoríase. Negava ainda antecedentes familiares ou farmacológicos relevantes. Encontrava-se medicada com naproxeno 500mg bid, com alívio parcial das queixas articulares. Ao exame objectivo apresentava lesões de lúpus discóide e telangiectasias na face (Fig. 1), sinovite dos punhos e da articulação tibiotársica direita e fenómeno de Raynaud em ambas mãos. Analiticamente destacava-se elevação da velocidade de sedimentação (26 mm/1ªh) e da fosfatase alcalina (128 U/L), sem evidência de citopenias ou de alteração da função renal. A electroforese das proteínas plasmáticas revelou hipergamaglobulinémia policlonal, com elevação das imunoglobulinas da classe M (IgM), 3,91 g/L. As serologias para sífilis, vírus da imunodeficiência humana, hepatite B e C foram negativas. O estudo de auto-imunidade revelou positividade para ANAs (Anti-Nuclear Antibodies; >1:1280), com padrão homogéneo (1:1280) e anti-centrómero (1:1280), bem como para anticorpos anti-dsDNA (double stranded DeoxyriboNucleicAcid; >48 UI/mL), anti-Ro/SSA (1:640) e anti-centrómero (1:1280) (Fig. 2). Apresentava anticorpos anticardiolipina e β2-glicoproteína da classe IgM em título alto, confirmado numa segunda determinação, bem como consumo de complemento CH50 (27,8 U/mL) e C3 (0,68 g/L). Verificou-se, ainda, positividade para AMAs (Anti-Mitochondrial Antibodies; 1:1280), com anticorpos anti-piruvato DH/IgG-M2 (>100U/mL), e para o factor reumatóide (FR; 62,2 UI/mL) com anticorpos anti-CCP (Cyclic Citrullinated Peptide) negativos. A pesquisa de ANCA (Anti-Neutrophil Cytoplasmic Antibody) e SACE (Serum Angiotensin Converting Enzyme), assim como o teste de anti-globulina directo foram negativos. Não apresentava proteinúria nas 24h ou evidência de serosite e/ou sinais indirectos de hipertensão pulmonar no ecocardiograma transtorácico. A radiografia das mãos apresentava ligeira osteopenia das articulações atingidas, sem evidência de erosões. Foi medicada com prednisolona 10mg/dia, com redução progressiva até 5mg/dia, e hidroxicloroquina 400mg/dia com melhoria significativa das queixas articulares. Iniciou ainda ácido ursodesoxicólico (10mg/Kg/dia) com normalização do valor de fosfatase alcalina. A capilaroscopia ungueal revelou, na mão direita, capilares dismórficos com aspecto de megacapilares, compatíveis com processo esclerodérmico (Fig. 3). A doente realizou estudo funcional respiratório, tomografia computorizada (TC) torácica de alta resolução e manometria esofágica que não evidenciaram alterações. A biópsia de glândula salivar minor revelou alterações histo-morfológicas compatíveis com Síndrome de Sjogrën (score de 2 no sistema de Greenspan). Após oito anos de evolução da doença, a doente desenvolveu alopecia difusa, não cicatricial com resolução após o aumento da dose de prednisolona para 40mg/dia e a associação de corticóide tópico em loção (butirato de hidrocortisona 1mg/mL).
Actualmente, ao fim de doze anos de doença, a doente cumpre critérios de classificação para quatro doenças auto-imunes diferentes, nomeadamente, Lúpus Eritematoso Sistémico (LES), Esclerose Sistémica (ES), Síndrome de Sjogrën (SS) e Colangite Biliar Primária (CBP). Apresenta 5 critérios da classificação ACR (American College of Rheumatology)/1997 para LES, nomeadamente rash discóide, fotossensibilidade, artrite, positividade para ANAs, anticorpos anti-dsDNA/anticardiolipina. Se se considera a classificação SLICC (Systemic Lupus International Collaborating Clinics)/2012 para LES, a doente cumpre 3 critérios clínicos (lúpus discóide, alopecia não cicatricial e artrite) e 4 critérios imunológicos (consumo de factores de complemento, positividade para ANAs, anticorpos anti-dsDNA e anticardiolipina). Apresenta 10 pontos na classificação ACR/EULAR (European League Against Rheumatism)/2013 de ES por apresentar telangiectasias (2 pontos), alterações na capilaroscopia ungueal (2 pontos), fenómeno de Raynaud (3 pontos) e anticorpos anti-centrómero (3 pontos). Cumpre dois critérios da classificação da SS ACR/2012, nomeadamente, positividade para anticorpos anti-Ro/SSA e sialadenite focal em biópsia de glândula salivar do lábio inferior. Por fim, apresenta ainda como critérios de diagnóstico de CBP, segundo a classificação da AASLD (American Association for the Study of Liver Diseases)/2009, evidência de colestase, com elevação da fosfatase alcalina, e positividade para AMAs.
A doente encontra-se medicada com deflazacort 6mg/dia, hidroxicloroquina 400mg/dia, ácido ursodesoxicólico 250mg bid e colecalciferol 28.000U mensalmente, mantendo seguimento semestral na consulta de doenças Auto-Imunes.
DISCUSSÃO
As síndromes de sobreposição (SSp) auto-imune definem-se pela presença de manifestações clínicas e/ou serológicas de duas ou mais doenças num mesmo indivíduo2. Conceito distinto, é o de doença indiferenciada do tecido conjuntivo que se define pela presença de manifestações clínicas e imunológicas sugestivas de doença do tecido conjuntivo específica sem que se cumpram os critérios de diagnóstico e/ou de classificação3. A prevalência estimada das SSp em doentes com doença do tecido conjuntivo é de 25%4, inferior à do LES e superior à da ES e das miosites inflamatórias. O espectro das SSp é heterogéneo, sendo possível o envolvimento de qualquer doença auto-imune. A sobreposição das diferentes doenças pode ocorrer em simultâneo ou, mais frequentemente, desenvolver-se ao longo de vários anos. A classificação das SSp é facilitada pela detecção de perfis de auto-anticorpos específicos, como acontece na doença mista do tecido conjuntivo na presença de anticorpos anti-nRNP (RiboNucleoProtein) e anti-p70, na síndrome anti-t-RNA (RiboNucleicAcid) sintetase com os anticorpos anti-Jo1 e anti-PL-7/12 e na escleromiosite associada aos anticorpos anti-PM/Scl e anti-Ku5. Há, contudo, SSp que carecem de um marcador serológico específico, como a síndrome de Rhupus, a associação LES/ES, AR/ES e AR/SS6.
L. Iaccarino et al. estimaram a prevalência da SS em doentes com LES em 14,8%5, sendo os marcadores imunológicos relacionados com a SS (anticorpos anti-Ro/SSA e anti-La/SSB, FR e hipergamaglobulinémia policlonal) mais frequentes em doentes com sobreposição LES/SS do que em doente com LES apenas e os anticorpos relacionados com o LES (ds-DNA, anti-Sm, anti-U1RNP e anti-cardiolipina) menos frequentes7. Os doentes com sobreposição LES/SS tendem a apresentar manifestações de LES de menor gravidade, como fadiga, fotossensibilidade, rash malar, úlceras orais, artrite e fenómeno de Raynaud8, sendo o risco de nefropatia menor7,9. Alguns estudos apontam para um maior risco de trombocitopenia nestes doentes7.
A prevalência estimada de SSp em doentes com ES, segundo L. Iaccarino et al., é de 16,2%, associando-se em 44,6% dos casos a DM/PM, 19,3% a AR, 18,5% a SS e 13,6% a LES5. A prevalência das formas limitada e difusa de ES nos indivíduos com SSp é de 83,6% e 16,4%, respectivamente, sendo o envolvimento pulmonar e a artrite mais frequentes do que em indivíduos com ES apenas10. Não tendo sido encontrado um marcador serológico específico da sobreposição LES/ES esta associa-se a uma elevada prevalência de anticorpos anti-dsDNA e anti-Scl70. A presença de anticorpos anti-centrómero foi documentada em 10,5% dos casos. Na sobreposição de ES/SS a prevalência de anticorpos anti-Ro/SSA e anti-La/SSB de 38,8% e 22,3%, respectivamente. A presença de anticorpos anti-centrómero é semelhante em doentes com ES e ES/SS, ao contrário, os anticorpos anti-Scl70 e anti-RNA polimerase são menos frequentes na sobreposição ES/SS.
Hernández-Molina et al. verificaram que os anticorpos anti-Ro/SSA e anti-La/SSB são mais frequentes em indivíduos com SS primário do que em doentes com sobreposição de outras doenças do tecido conjuntivo11, contudo, nestes parece associar-se a formas mais severas de síndrome seca e tumefacção parotídea12. Os mesmos autores encontraram uma prevalência semelhante de infiltração linfocitária em biópsia de glândula salivar em ambos grupos.
A CBP é considerada por alguns autores uma patologia de sobreposição entre a patologia auto-imune hepática e reumatológica. Floreani et al. encontraram uma prevalência de doença auto-imune extra-hepática de 61,2% numa população de doentes com CBP13. A SS é a doença auto-imune que mais frequentemente se associa a CBP, seguida da forma limitada de ES, a síndrome de CREST (Calcinosis Raynauds Esophageal-dysfunction Sclerodactyly Telangectasias) e da tiroidite de Hashimoto. São raros os casos descritos de sobreposição com LES14. A associação à ES parece relacionar-se com uma progressão mais lenta para cirrose15. Telfer Reynolds descreveu pela primeira vez a rara associação de ES e CBP, pelo que ficou conhecida como síndrome de Reynolds.
A terapêutica das SSp tem por base a corticoterapia e fármacos imunossupressores, dirigindo-se em cada momento à manifestação dominante. O recurso à terapêutica biológica, com agentes anti-TNFα (Tumor Necrosis Factor α) e anti-CD20 (Cluster Diferentiation 20), tem ocorrido nos casos de doença refractária5.
CONCLUSÃO
A sobreposição de perfis de auto-anticorpos, bem como de distintos fenótipos de diferentes doenças num mesmo indivíduo, representam, para o clínico, um desafio no diagnóstico, seguimento e tratamento destes doentes.Figura I
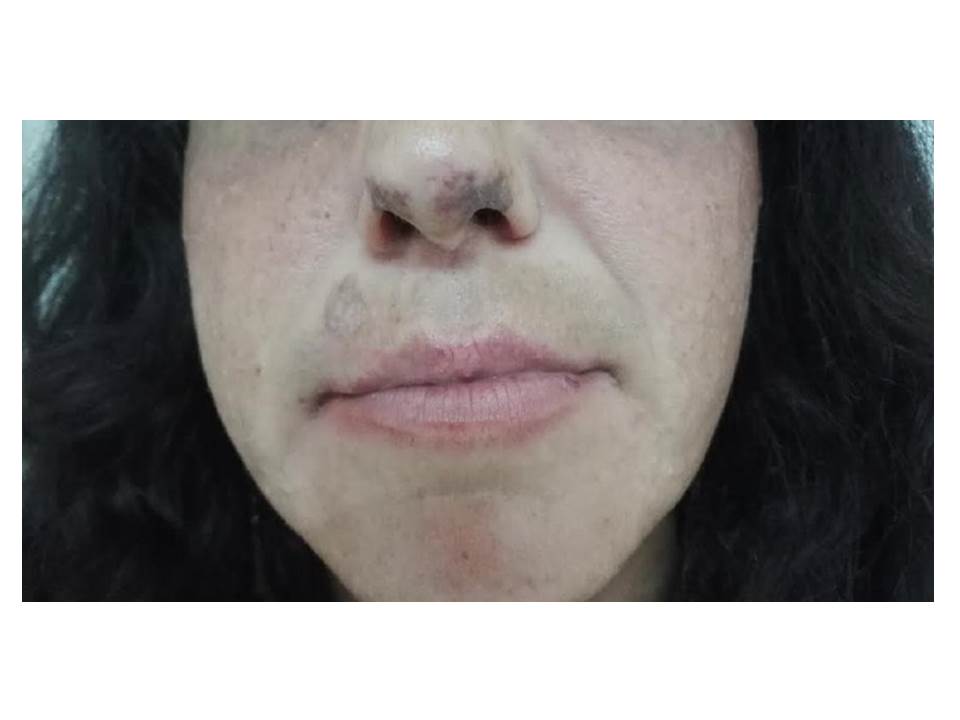
Lesões de lúpus discóide na face e na ponta do nariz; telangiectasias na mucosa do lábio inferior.
Figura II
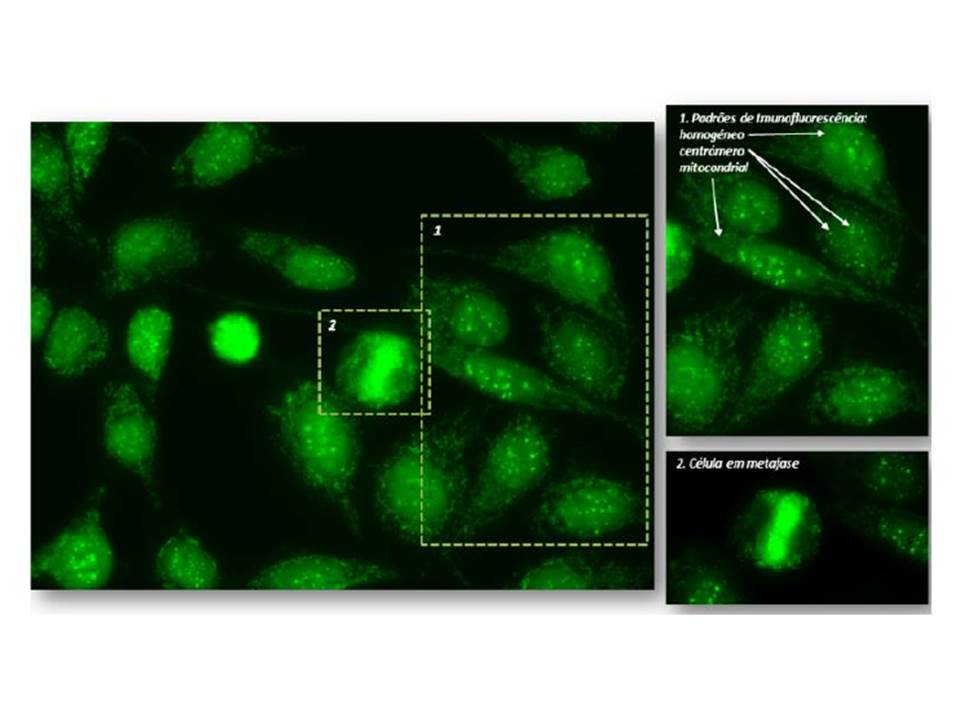
Padrões de imunofluorescência indirecta (células Hep2). Em células em interfase, observam-se os padrões nucleares homogéneo e centrómero, assim como o padrão citoplasmático mitocondrial. Na 2ª ampliação, célula em mitose com fluorescência granular múltipla alinhada na placa metafásica (diferente ponto de focagem para melhor visualização).
Figura III
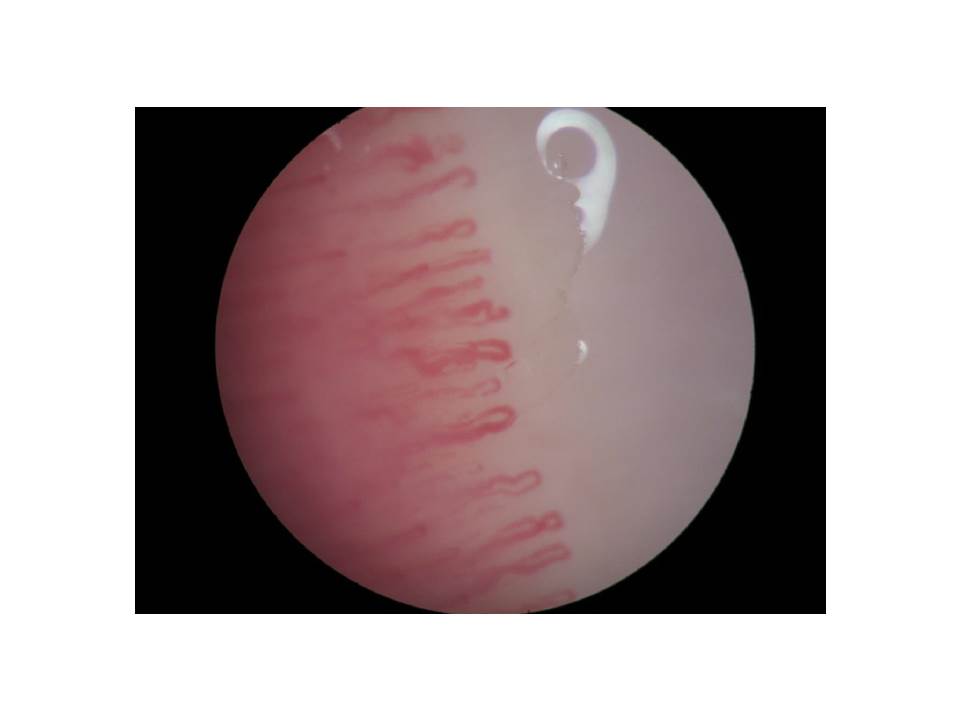
Imagem de capilaroscopia ungueal. Evidência de múltiplos capilares dilatados e presença de megacapilares, ainda sem áreas avasculares significativas, alterações a sugerir padrão esclerodérmico precoce/activo.
BIBLIOGRAFIA
1. Jury E, DCruz D, Morrow W. Autoantibodies and overlap syndromes in autoimmune rheumatic disease. J Clin Pathol. 2001;54:340347.
2. Fine RM. Overlap syndromes. Int J Dermatol. 1991;30:479-480.
3. Mosca M, Neri R, Bombardieri S. Undifferentiated connective tissue diseases (UCTD): a review of the literature and a proposal for preliminary classification criteria.Clin Exp Rheumatol. 1999;17(5):615-620.
4. Cervera R, Khamashta MA, Hughes GVR. Overlap syndromes. Ann Rheum Dis. 1990;49:947948.
5. Iaccarino L,Gatto M,Bettio S,Caso F,Rampudda M,Zen M,et al. Overlap connective tissue disease syndromes. Autoimmun Rev.2013;12(3):363-73.
6. Rekvig OP, Putterman C, Casu C, Gao HX, Ghirardello A, Mortensen ES, et al. Autoantibodies in lupus: culprits or passive bystanders?. Autoimmun Rev. 2012;11:596603.
7. Manoussakis MN, Georgopoulou C, Zintzaras E, Spyropoulou M, Stavropoulou A, Skopouli FN, et al. Sjögren´s syndrome associated with systemic lupus erythematosus: clinical and laboratory profiles and comparison with primary Sjögren´s syndrome. Arthritis Rheum. 2004;50:882891.
8. Baer AN, Maynard JW, Shaikh F, Magder LS, Petri M. Secondary Sjögren´s syndrome in systemic lupus erythematosus defines a distinct disease subset. J Rheumatol. 2010;37:11431149.
9. Osio-Salido E, Manapat-Reyes H. Epidemiology of systemic lupus erythematosus in Asia. Lupus. 2010;19:13651373.
10. Hunzelmann N, Genth E, Krieg T, Lehmacher W, Melchers I, Meurer M, et al. The registry of the German Network for Systemic Scleroderma: frequency of disease subsets and patterns of organ involvement. Rheumatology (Oxford). 2008;47:11851192.
11. Hernández-Molina G, Avila-Casado C, Cárdenas-Velázquez F, Hernández-Hernández C, Calderillo ML, Marroquín V, et al. Similarities and differences between primary and secondary Sjögren´s syndrome. J Rheumatol. 2010;37:800808.
12. Droxos A, Andonopoulos AP, Costopoulos JS, Stavropoulos ED, Papademetriou CS, Moutsopoulos HM. Sjogren´s syndrome in progressive systemic sclerosis. J Rheumatol. 1988;15:965-969.
13. Floreani A, Franceschet I, Cazzagon N, Spinazzè A,Buja A,Furlan P, et al. Extrahepatic autoimmune conditions associated with primary biliary cirrhosis.Clin Rev Allergy Immunol. 2015;48(2-3):192-197.
14. Efe C, Purnak T, Ozaslan E, Ozbalkan Z. The importance of autoimmune hepatitis and primary biliary cirrhosis in patients with systemic lupus erythematosus.Lupus. 2011;20:112.
15. Rigamonti C, Shand LM, Feudjo M,Bunn CC, Black CM, Denton CP, et al. Clinical features and prognosis of primary biliary cirrhosis associated with systemic sclerosis.Gut. 2006;55:388394.