

Introdução:
O aumento de espessura da parede do ventrículo esquerdo (VE) pode dever-se a sobrecarga de pressão, hipertrofia dos miócitos de causa genética ou ao depósito de substâncias anómalas intra ou extra-celulares. Apesar de poder ser assintomática, surgindo como um achado num exame complementar, é essencial efectuar o diagnóstico etiológico pelas implicações terapêuticas e prognósticas. Apresenta-se um caso em que apenas a investigação completa e sistematizada permitiu obter o diagnóstico.
Caso clínico:
Homem caucasiano de 60 anos, assintomático, foi observado em consulta de Cardiologia para controlo do risco cardiovascular. Tinha antecedentes de hipertensão arterial >10 anos, controlada com valsartan+hidroclorotiazida 160mg/12,5mg e lercanidipina 10mg, hipercolesterolémia sob rosuvastatina 10mg, anomalia da glicémia em jejum, obesidade grau 1 e tabagismo prévio, em evicção há 20 anos. Na história familiar não havia referência a doença cardíaca ou morte súbita.
Na observação destacava-se: pressão arterial 138/81mmHg, frequência cardíaca 60 bpm e sopro sistólico grau II/VI, apical, com irradiação axilar. No electrocardiograma (ECG) apresentava ritmo sinusal, frequência de 55 bpm, desvio esquerdo do eixo eléctrico, alterações inespecíficas da repolarização ventricular, sem critérios de voltagem para hipertrofia ventricular esquerda (HVE). Analiticamente sem alterações relevantes.
Para esclarecimento do sopro cardíaco realizou ecocardiograma transtorácico (ETT) que mostrou ventrículo esquerdo de cavidade pequena, com hipertrofia concêntrica de todas as paredes (HVE simétrica), máxima ao nível do septo interventricular (Fig.1-A) (21mm referência <10 mm);1 presença de movimento sistólico anterior (SAM) do folheto anterior da válvula mitral, sem obstrução significativa (gradiente máximo intraventricular de 22mmHg em repouso); disfunção diastólica grau I, provável elevação das pressões de enchimento ventricular e dilatação auricular esquerda (48ml/m2 referência <34 ml/m2).1
Com o objectivo de caracterizar a HVE realizou ressonância magnética cardíaca (RM-C) que mostrou um padrão de realce tardio intramiocárdico a nível basal e mediano na parede ínfero-lateral, sugestivo de doença de Anderson-Fabry (Fig.1-B). Esta suspeita excluiu-se pelo doseamento normal de α-galactosidase. Nesta faixa etária, com HVE simétrica e RM-C sugestiva de infiltração, optou-se por descartar amiloidose através de imunofixação de imunoglobulinas no sangue e urina, que foi negativa, e biópsia de gordura abdominal que revelou tecido adiposo sem substância amilóide. Perante os resultados negativos, sabendo-se da existência de formas de deposição miocárdica preferencial, efectuou-se biópsia endomiocárdica do VD e também do VE. Histologicamente havia fibrose intersticial e hipertrofia miocitária sem padrão específico, sem depósito de substâncias anómalas, sinais inflamatórios ou neoplásicos. (Fig.1-C e D)
Mantendo-se a dúvida quanto à etiologia, pediu-se estudo genético das proteínas sarcoméricas que não revelou mutações patogénicas mas identificou variantes genéticas rs2303510 e rs45533739 em homozigotia nos genes FHOD3 e TNNT2, respectivamente, que aumentam a susceptibilidade para miocardiopatia hipertrófica (MCH).2,3
Fez-se então o diagnóstico provável de MCH sarcomérica de instalação tardia, tendo-se procedido à estratificação de risco arrítmico do doente. Foi proposto rastreio aos familiares, que, mesmo depois de esclarecidos sobre a sua importância, o recusaram.
Discussão
A classificação das miocardiopatias tem sido alvo de várias actualizações que acompanham os avanços na compreensão dos seus mecanismos fisiopatológicos. Em 2008 a Sociedade Europeia de Cardiologia propôs um sistema de classificação baseado na morfologia e função do VE, independentemente da etiologia,4 que se manteve nas recomendações de 2014.5 Nesses documentos, a MCH é definida como um aumento da espessura das paredes do VE>15mm no adulto, não explicável por condições de sobrecarga. Esta definição inclui os casos de hipertrofia dos miócitos de causa genética, bem como algumas situações de infiltração intersticial ou acumulação de substâncias intracelulares condicionando aumento da espessura ventricular.
Esta abordagem implica excluir situações de sobrecarga de pressão ventricular que originem hipertrofia miocárdica compensatória. As mais frequentes nos adultos são a hipertensão arterial e a estenose aórtica, podendo também ocorrer em atletas de alta competição. Neste caso, o diagnóstico diferencial colocou-se com a cardiopatia hipertensiva, embora a espessura parietal>15mm favorecesse o diagnóstico de MCH.5
Cerca de 60% dos casos de MCH surgem por mutações autossómicas dominantes nos genes das proteínas sarcoméricas, denominando-se MCH sarcomérica5 e afectando 1/500 indivíduos da população.6 Menor percentagem (5-10%) deve-se a um conjunto de doenças infiltrativas, metabólicas, endocrinológicas, neuromusculares, mitocondriais ou síndromes malformativos. Infelizmente não é possível chegar ao diagnóstico etiológico em cerca de 25-30% dos casos. Quando diagnosticada em adulto, a lista de causas não relacionadas com as proteínas sarcoméricas diminui, incluindo fundamentalmente a doença de Fabry e a amiloidose cardíaca. Ainda assim, as diferentes etiologias têm significados distintos quanto a tratamento, prognóstico e implicações familiares, pelo que a suspeita de MCH deve desencadear a procura da causa subjacente, seguindo uma abordagem diagnóstica faseada. (Fig 2)
A avaliação do doente começa na história clínica. Muitos portadores de MCH são assintomáticos, sendo a sua detecção um achado fortuito. Quando sintomáticos, podem referir angor de esforço ou em repouso, palpitações, sintomas de insuficiência cardíaca ou síncope.5 Nesta avaliação deve ser incluída a história familiar, identificando os casos de morte súbita, transplante cardíaco, implantação de pacemaker ou cardiodesfibrilhador, sintomas de insuficiência cardíaca inexplicável ou doenças sistémicas associadas a MCH.5
O exame físico pode igualmente ser inocente ou identificar um sopro sistólico de ejecção típico de obstrução do tracto de saída VE - no bordo esquerdo do esterno, irradiando ao ápex, que aumenta de intensidade com manobra de Valsalva. É igualmente importante identificar sintomas e sinais extra-cardíacos que sugiram doenças específicas não-sarcoméricas como por exemplo surdez, cataratas, dor neuropática e angioqueratomas (na doença de Fabry) ou síndrome nefrótico, síndrome do canal cárpico, macroglossia, púrpura peri-orbitária e parestesias (na amiloidose).7,8
No electrocardiograma, embora com baixa sensibilidade, as alterações mais frequentes são o aumento de voltagem do QRS, desvio esquerdo do eixo eléctrico e aumento da duração do QRS. Pode ainda existir inversão profunda e simétrica das ondas T precordiais quando a HVE é sobretudo apical; baixa voltagem é um achado paradoxal e sugere amiloidose.5
A ecocardiografia, essencial na abordagem diagnóstica, avalia a espessura parietal e sua distribuição e identifica a presença de obstrução dinâmica do tracto de saída do VE por SAM da válvula mitral. Esta obstrução, não sendo patognomónica, está presente em repouso em um terço dos doentes com MCH e noutro terço durante manobras que aumentam a sobrecarga ventricular (Valsalva). Determinadas características ecocardiográficas podem evocar doenças específicas: HVE assimétrica sugere MCH sarcomérica, enquanto a simétrica é mais comum em doenças metabólicas e infiltrativas; o aspecto granulado do miocárdio, a hipertrofia do septo inter-auricular e derrame pericárdico são sugestivos de amiloidose. No presente caso, o ecocardiograma foi equívoco pois a hipertrofia afectava todas as paredes. Por este motivo optou-se pela realização de RM-C, exame que deve ser considerado nos casos de MCH em que o ecocardiograma deixa dúvidas. O realce tardio com gadolíneo pode apresentar padrões característicos, fornecendo pistas para o diagnóstico etiológico: na MCH sarcomérica está habitualmente presente nas áreas de hipertrofia e nos pontos de inserção anterior e posterior do VD; na doença de Fabry é mais frequentemente observado na porção basal da parede infero-lateral; na amiloidose assume uma distribuição difusa subendocárdica.9 No caso apresentado, o padrão era sugestivo de doença de Fabry, que não se confirmou.
Na marcha diagnóstica da MCH, os testes laboratoriais destinam-se sobretudo à exclusão de doenças específicas. A doença de Fabry é diagnosticada pelos níveis reduzidos da enzima α-galactosidase A e, nas mulheres, também pela pesquisa de mutações no gene GLA (Xq22).10 A imunofixação sérica e urinária de imunoglobulinas é útil no diagnóstico de amiloidose primária, devendo ser confirmado através de biópsia. O local mais acessível é a gordura abdominal positiva em 65-95% dos casos seguindo-se as glândulas salivares e recto, ou a biópsia do órgão afectado se estas forem inconclusivas e a suspeita se mantiver.8 Efectivamente, a biópsia endomiocárdica está recomendada quando há suspeita de doença infiltrativa, inflamatória ou de armazenamento impossível de confirmar por outros métodos.5 Foi o caso deste doente.
Porque a biópsia revelou hipertrofia miocitária inespecífica, compatível com etiologia sarcomérica, e porque o doente tinha descendência em idade fértil, optou-se pela realização do teste genético que identificou polimorfismos facilitadores do aparecimento de MCH.2,3 O papel do teste genético permanece ainda sob algum debate: por um lado o seu custo impede a realização em todos os doentes com diagnóstico clínico de MCH, por outro lado a identificação da mutação específica permite confirmar o diagnóstico no caso-índice e rastrear casos pré-sintomáticos entre os familiares evitando-se vigilância clínica prolongada em muitos indivíduos. Assim, as orientações europeias recomendam o teste genético nos doentes que preenchem os critérios de MCH quando este permite o rastreio e aconselhamento dos familiares.5
Ainda que muitos doentes com MCH sarcomérica sejam assintomáticos, apresentam risco aumentado de insuficiência cardíaca, arritmias, síncope e morte súbita. Recomenda-se o seguimento regular observação clínica, ECG e ETT anual ou bianual - focado na detecção precoce de disfunção VE, obstrução do tracto de saída VE e arritmias. Com base nessa avaliação, os doentes podem ser candidatos a terapêutica médica ou colocação de cardioversor-disfibrilhador implantável, para prevenção de morte súbita cardíaca.5 Por outro lado, o aparecimento recente de novas terapêuticas dirigidas ao gene mutado ou às alterações moleculares responsáveis pela doença (sensibilidade das miofibrilhas ao cálcio, desregulação dos canais iónicos, etc.)11 constituem uma nova estratégia de prevenção que torna ainda mais premente o diagnóstico precoce.
Figura 1: Exames complementares de diagnóstico
A - Ecocardiograma transtorácico Ventrículo esquerdo em plano apical quatro câmaras
B - Ressonância magnética cardíaca padrão de realce tardio com gadolínio ( → )
C - Biópsia Endomiocárdica: Hematoxilina eosina - Miócitos hipertrofiados ( →), Fibrose (*).
D -Biópsia Endomiocárdica: Tricómico de Masson - Miócitos hipertrofiados ( → ) Fibrose (*).
Figura 2 -Algoritmo de abordagem diagnóstica da Hipertrofia Ventricular esquerda (adaptado de Elliot et al.5)
Figura I
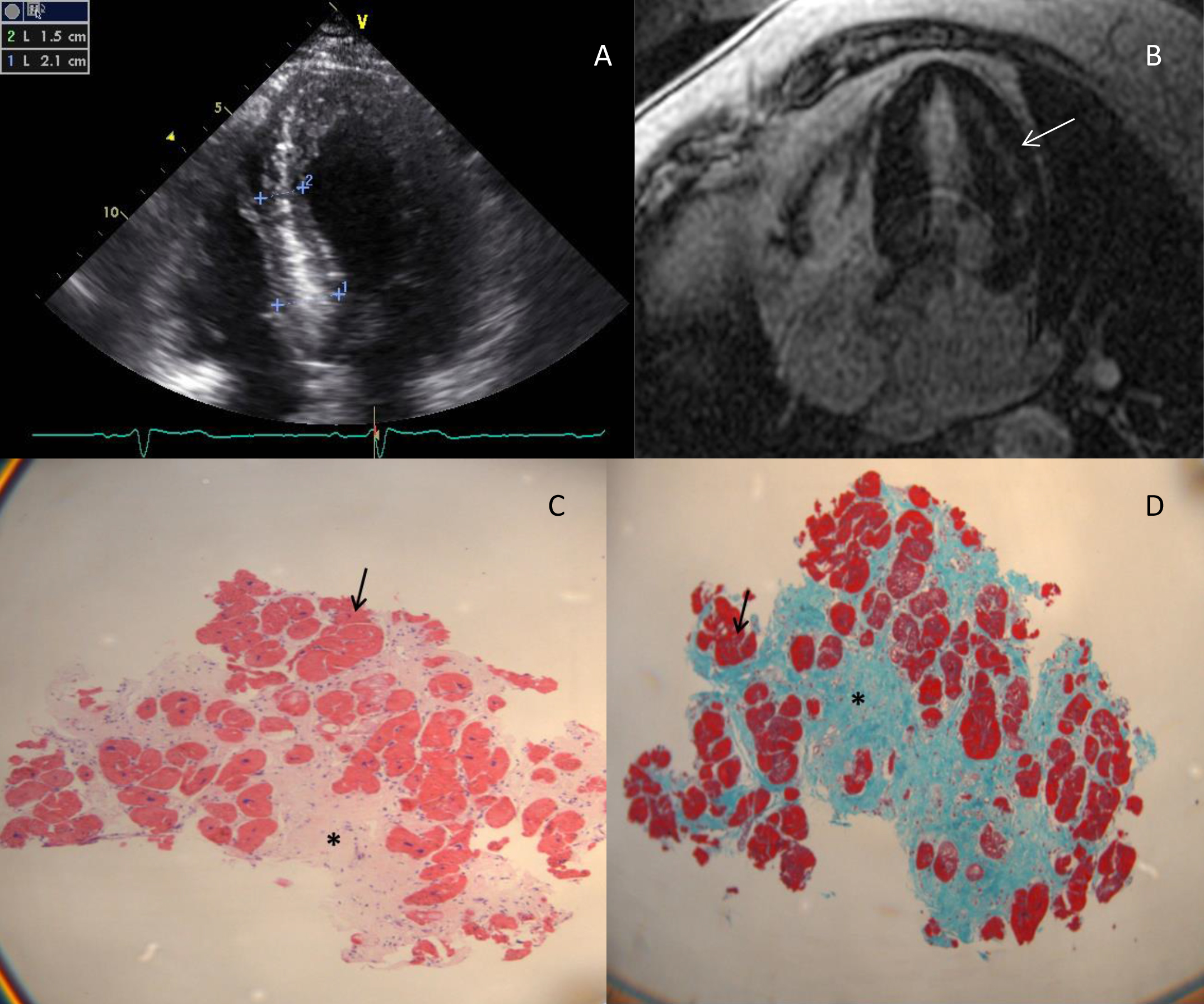
Figura 1: Exames complementares de diagnóstico A - Ecocardiograma transtorácico Ventrículo esquerdo em plano apical quatro câmaras B - Ressonância magnética cardíaca padrão de realce tardio com gadolínio ( → ) C - Biópsia Endomiocárdica: Hematoxilina eosina - Miócitos hipertrofiados ( → ), Fibrose (*). D -Biópsia Endomiocárdica: Tricómico de Masson - Miócitos hipertrofiados ( → ) Fibrose (*).
Figura II
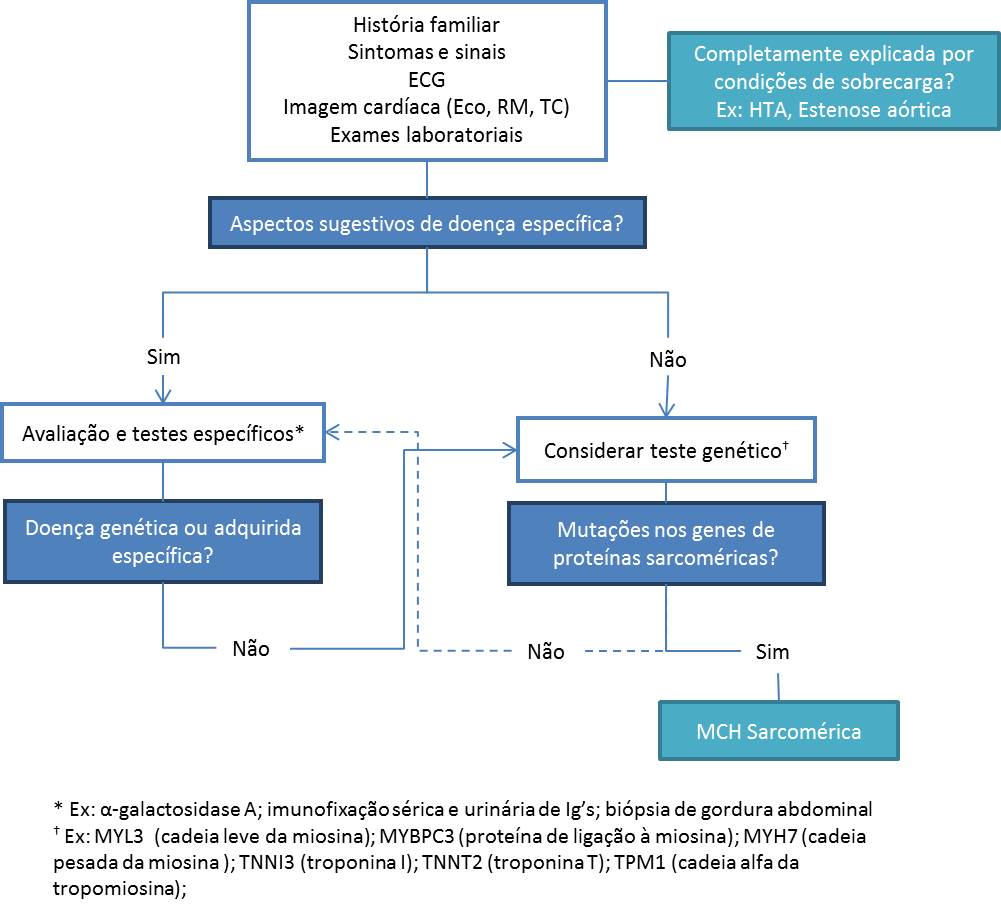
Algoritmo de abordagem diagnóstica da Hipertrofia Ventricular esquerda (adaptado de Elliot et al.5)
BIBLIOGRAFIA
Referências:
1. Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American society of echocardiography and the European association of cardiovascular imaging. Eur Heart J Cardiovasc Imaging. 2015;16(3):23371.
2. Wooten EC, Hebl VB, Wolf MJ, Greytak SR, Orr NM, Draper I, et al. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2013;6(1):108.
3. Jáchymová M, Muravská a., Paleček T, Kuchynka P, Řeháková H, Magage S, et al. Genetic variation screening of TNNT2 gene in a cohort of patients with hypertrophic and dilated cardiomyopathy. Physiol Res. 2012;61(2):16975.
4. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2007;29(2):2706.
5. Elliott PM, Anastasakis a., Borger M a., Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;273379.
6. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults : Echocardiographic Analysis of 4111 Subjects in the CARDIA Study. Circulation. 1995 Aug 15;92(4):7859.
7. Rapezzi C, Arbustini E, Caforio ALP, Charron P, Gimeno-Blanes J, Heliö T, et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34(19):144858.
8. Seldin DC, Berk JL, Sam F, Sanchorawala V. Amyloidotic Cardiomyopathy: Multidisciplinary Approach to Diagnosis and Treatment. Heart Fail Clin. 2011;7(3):38593.
9. Mahrholdt H, Wagner A, Judd RM, Sechtem U, Kim RJ. Delayed enhancement cardiovascular magnetic resonance assessment of non-ischaemic cardiomyopathies. Eur Heart J. 2005;26(15):146174.
10. Correia E, Vidinha J, Rodrigues B, Santos L, Moreira D, Garrido J, et al. [Description of a new mutation in a female patient with Fabry disease]. Rev Port Cardiol orgão Of da Soc Port Cardiol = Port J Cardiol an Off J Port Soc Cardiol. 2011 Oct 1;30(10):78993.