O Feocromocitoma é um tumor raro, neuroendócrino, secretor de catecolaminas responsável por hipertensão arterial potencialmente fatal. As suas manifestações multissistémicas que mimetizam outros diagnósticos, fazem desta entidade um verdadeiro desafio clínico.
O diagnóstico depende da confirmação bioquímica da sobreprodução de catecolaminas e metanefrinas e da localização anatómica através de métodos de imagem, sendo a ressecção cirúrgica o seu tratamento definitivo com cuidados pré-operatórios específicos [1-3].
Os autores apresentam o caso de um doente do sexo masculino de 66 anos, leucodérmico, com história de Hipertensão Arterial (HTA), Diabetes mellitus não insulinotratada e insuficiência renal, que deu entrada no Serviço de Urgência com mal-estar geral, náuseas e vómitos, parestesias, mialgias e palpitações.
Ao exame objectivo constatou-se taquicardia sinusal (107 bpm), sem outras alterações relevantes. A avaliação analítica revelou rabdomiólise, lesão renal aguda, hiperglicémia e leucocitose sem neutrofilia ou linfocitose.
O doente foi admitido na enfermaria de Medicina Interna para tratamento e investigação etiológica.
Durante o internamento, verificou-se instabilidade hemodinâmica com frequências cardíacas elevadas (120 bpm), períodos de hipertensão (200/100mmHg) alternando com hipotensão (80/52 mmHg), confusão, ansiedade, palpitações, náuseas e vómitos.
Foi detectada a presença de metanefrinas fraccionadas em exame de urina/24 horas com metanefrinas = 5.985 mcg (<0.400), normetanefrinas e catecolaminas dentro dos valores de referência, com posterior confirmação imagiológicade uma massa suprarrenal volumosa(TAC abdominal figura 1ecintigrafia com123I-MIBG).
Após o diagnóstico, o doente foi submetido a fluidoterapia vigorosa e controlo da HTA com duplo bloqueio de receptores α (Fenoxibenzamina) e β (Nebivolol), sendo posteriormente admitido para cirurgia com ressecção da massa por via laparotómica (figura 2).
Após remoção do tumor houve melhoria clínica global, com remissão dos sintomas, normalização dos valores de aminas e resolução completa da HTA, hiperglicémia e insuficiência renal.
O Feocromocitoma é uma entidade rara cujo diagnóstico é de fundamental importância no sentido de prevenir a ocorrência de eventos com alta morbilidade e mortalidade. A sua suspeição atempada, com abordagem terapêutica médica (bloqueio adrenérgico e hidratação) e cirúrgica (via laparoscópica ou laparotómica) dirigidas são fulcrais para o prognóstico destes doentes [1].
Figura I
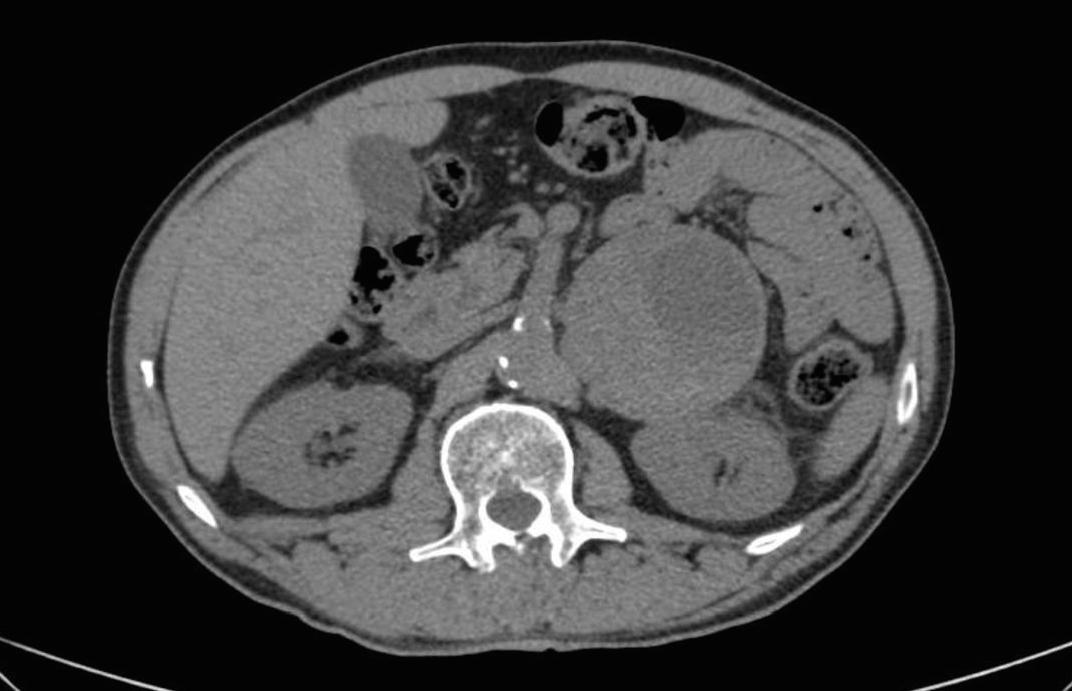
Volumosa massa suprarrenal esquerda observada na TAC abdominal
Figura II
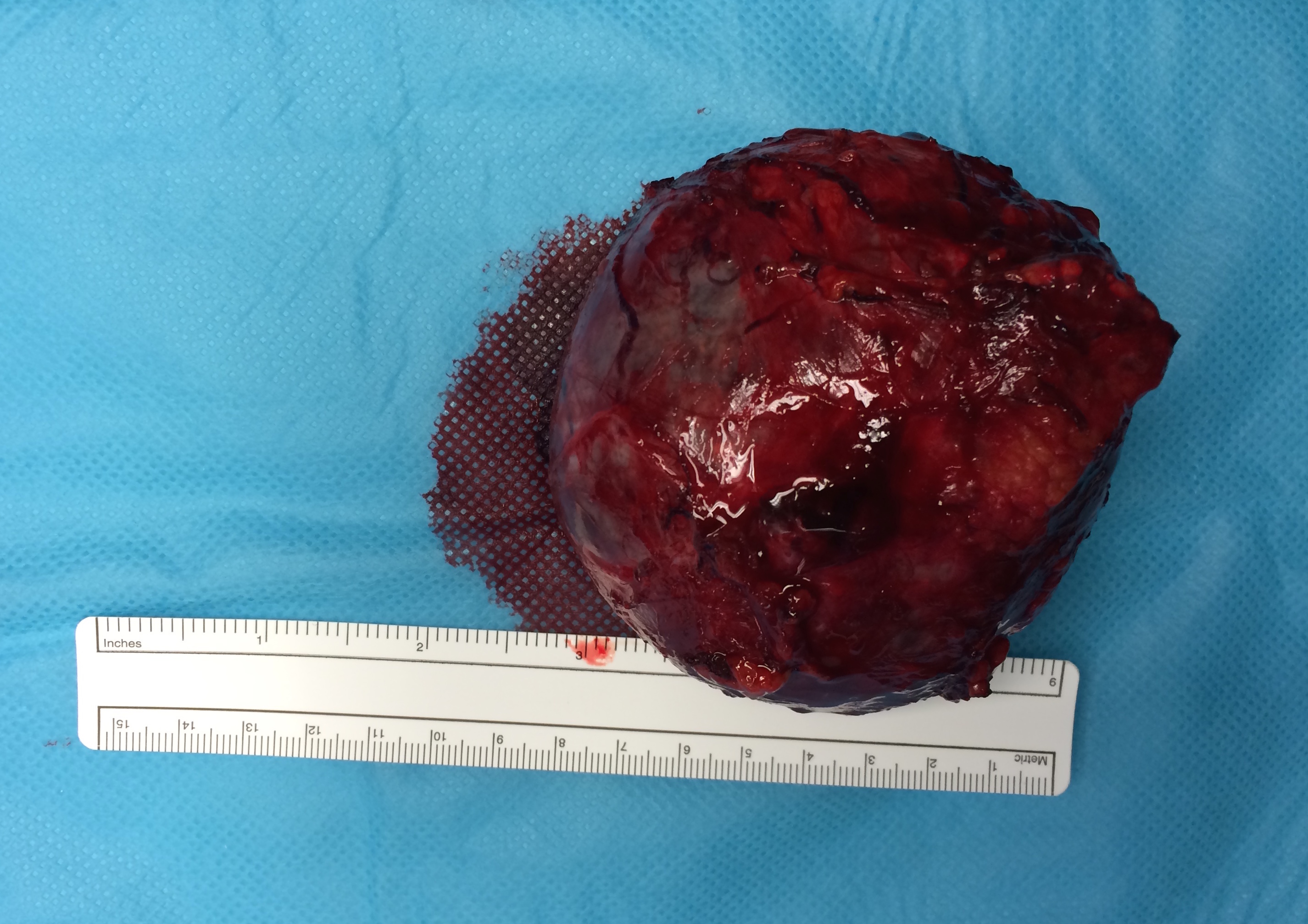
Aspecto macroscópico da massa suprarrenal após ressecção via laparotómica com dimensões 7.5x7.8x6.3 cm e peso de 152g (microscopicamente: presença de cápsula, pleomorfismo ligeiro a moderado, índice Ki-67 < 3% e positividade para cromogranina, sinaptofisina e pS100)
BIBLIOGRAFIA
1. Jacques W. M. Lenders, Quan-Yang Duh, Graeme Eisenhofer, Anne-Paule Gimenez-Roqueplo, Stefan K. G. Grebe, Mohammad Hassan Murad, et al. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, June 2014, 99(6):19151942
2. Ibrahim M. Zardawi. Phaeochromocytoma masquerading as anxiety and depression. Am J Case Rep 2013; 14:161-163
3. Jean-Philippe Baguet, Laure Hammer, Tânia Longo Mazzuco, Olivier Chabre, Jean-Michel Mallion, Nathalie Sturm, et al. Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. European Journal of Endocrinology (2004) 150 681686

