INTRODUÇÃO:
A hipertensão pulmonar (HP) é um estado hemodinâmico que pode ocorrer de forma isolada ou associada a diferentes entidades clínicas e que se caracteriza pela presença de uma pressão arterial pulmonar média (PAPm) igual ou superior a 25mmHg em repouso e avaliada por cateterismo cardíaco direito1,2,3. A sua prevalência estimada é de 97 casos/milhão de pessoas, com predomínio nas mulheres (1.8:1)1. A hipertensão arterial pulmonar idiopática (HAPi) caracteriza-se por HP com uma pressão de oclusão da artéria pulmonar (POAP) inferior ou igual a 15mmHg e resistências vasculares pulmonares (RVP) superiores a 3WU, na ausência de outras causas de HP1. A HAPi, um diagnóstico de exclusão, é uma doença rara, de etiologia desconhecida, com comportamento maligno e sobrevida curta, média de 2.8 anos se não tratada adequadamente2. Tem uma prevalência estimada de 5.9 casos/milhão de pessoas, sendo a maioria dos casos diagnosticada entre os 50 e os 60 anos1,4.
CASO CLÍNICO:
Um homem de 55 anos, com antecedentes pessoais de adenocarcinoma do cólon, tratado com hemicolectomia e em remissão clínica há 4 anos, columbófilo desde a juventude e sem história de consumo de drogas ou tóxicos, recorreu a consulta de especialidade por dispneia para médios esforços de aparecimento recente; na altura não apresentava ortopneia, DPN, tosse, pieira ou dor torácica. Por apresentar estudo positivo para precipitinas séricas para penas de pombos, e sem estudo funcional, imagiológico ou do pulmão profundo por lavagem bronchoalveolar que suportassem um diagnóstico de pneumonite de hipersensibilidade foi-lhe instituída terapêutica com broncodilatadores e prednisolona oral (1mg/kg/dia) e evicção do contacto com os pombos. Por roncopatia, realizou estudo do sono que mostrou índice de apneia/hipopneia 50/h, pelo que iniciou auto-CPAP domiciliário.
Apesar das medidas tomadas a evolução foi negativa com agravamento progressivo da dispneia de esforço pelo que 1 mês depois realiza ecocardiograma transtorácico (ETT) que mostrou dilatação das cavidades cardíacas direitas e hipertensão pulmonar severa, com PSAP estimada em 90mmHg. Os exames complementares realizados nessa altura revelaram: gasometria arterial sem insuficiência respiratória; radiografia de tórax com engurgitamento hilar bilateral, ICT > 0,5 e alargamento do bordo direito da silhueta cardíaca (figura 1); a TAC pulmonar com angiografia não revelou alterações parenquimatosas nem evidência de sinais de tromboembolismo pulmonar, confirmando sinais de hipertensão pulmonar e aumento das dimensões das câmaras cardíacas direitas; o ECG: ritmo sinusal, frequência cardíaca 70bpm; onda p pulmonar e bloqueio incompleto de ramo direito (figura 2). Foi mantida a terapêutica com broncodilatadores e auto-CPAP. Por agravamento da dispneia, agora com dispneia em repouso, e aparecimento de edema simétrico dos membros inferiores é hospitalizado. Ao exame objetivo, estava polipneico em repouso, com tiragem supra-clavicular e cianose labial. Encontrava-se hemodinamicamente estável, com taquicardia sinusal e a auscultação cardíaca e pulmonar não revelavam alterações, exceto uma acentuação do 2º som cardíaco no foco pulmonar. Apresentava edemas bimaleolares, bilaterais, hipocratismo digital. A gasometria, em ar ambiente, revelou uma insuficiência respiratória tipo 1 (ph de 7.52, pO2 de 55 mmHg, pCO2 de 26 mmHg, HCO3- de 22 mEq/l e Sat O2 91%) e a análise dos exames já efetuados (ECG, Radiografia de tórax, ETT e TAC torácico com angiogragia) confirmou a suspeita de hipertensão pulmonar como causa para o quadro clínico do doente. Foi complementado o estudo prévio com: estudo analítico (hemograma, bioquímica, função tiroideia, serologias víricas para o VIH, hepatite B e C e estudo imunológico -ANAs, Ac anti-centrómero, anti-RNP, anti-Scl70, FR-) que foi negativo, BNP que estava ligeiramente elevado (260UI/L); provas funcionais respiratórias revelaram curvas débito/volume normais e DLCO/VA reduzida (56%); cintigrafia pulmonar de ventilação-perfusão que não apresentava alterações da ventilação ou perfusão e ecografia abdominal que foi normal e sem evidência de hipertensão portal.
O cateterismo cardíaco direito (CCD) confirmou uma hipertensão pulmonar pré-capilar (PAPm de 45mmHg com PAOP de 11mmHg), com critérios de gravidade (pressão aurícula direita de 13mmHg e índice cardíaco de 1,26l/min/m2); as resistências vasculares pulmonares estavam elevadas (13.5UW) e o teste de vasoreatividade aguda pulmonar (TVR) foi negativo.
Confirmado o diagnóstico de HAPi foi proposto e aceito no Centro oficial de Tratamento da HP para a Região Norte.
Aí, assumido o diagnóstico de HAPi com TVR negativo e com critérios de gravidade (evolução clínica rapidamente progressiva, em classe funcional IV, incapaz de efetuar teste de marcha, com NT-proBNP elevado, pressão da aurícula direita elevada e índice cardíaco baixo). Iniciou terapêutica específica tripla com bosentano, sildenafilo e iloprost inalado.
Aos 3 meses de tratamento, apresentava franca melhoria clínica (Tabela 1). Aos 12 meses houve ligeiro agravamento clínico (classe funcional III, agravamento da insuficiência respiratória, diminuição da distância percorrida no teste de 6 minutos de marcha (T6MM), subida de NT-proBNP e persistência dos critérios hemodinâmicos de gravidade), pelo que iniciou epoprostenol endovenoso contínuo, com aumento progressivo da dose até aos 20ng/Kg/min, em substituição do iloprost inalado. Posteriormente e para melhorar a comodidade da terapêutica foram substituídos o bosentano e o sildenafilo por fármacos do mesmo grupo, macitentano e tadalafilo, respetivamente.
A evolução clínica tem sido favorável, apesar de intercorrência grave (abdómen agudo por oclusão intestinal por brida que obrigou a cirurgia de urgência), com melhoria progressiva de todos os marcadores de prognóstico da doença. À data da última avaliação (Julho de 2016), mantinha-se em classe funcional II, capaz de exercício físico de intensidade moderada (caminhada diária e praticando danças de salão), com um bom T6MM mas ainda com pro-BNP ligeiramente elevado, pelo que se encontra em ajuste progressivo da dose de epoprostenol (atualmente com 25ng/Kg/min). Mantém oxigenoterapia de longa duração.
DISCUSSÃO:
A HAPI é uma doença crónica, com comportamento maligno, caracterizada por uma remodelação progressiva da vasculatura pulmonar que conduz ao aumento da resistência vascular pulmonar, insuficiência cardíaca direita e morte.5 No entanto, apesar da ausência de cura, as terapêuticas atualmente disponíveis permitem atrasar a evolução da doença, melhorar os sintomas e aumentar a funcionalidade e qualidade de vida dos doentes6. Nos últimos 30 anos, foram descobertos um número apreciável de fármacos (10 com eficácia demonstrada) e testadas diversas estratégias terapêuticas (combinações de fármacos das diferentes classes) que nos permitem encarar o futuro com otimismo5. Uma meta-análise de 23 ensaios clínicos demonstrou a redução da mortalidade com as terapêuticas aprovadas no tratamento dos doentes com HAP7. Deve ser tido em conta que o diagnóstico e tratamento precoces influenciam significativamente os resultados da terapêutica e o prognóstico da doença. A classe funcional, a distância percorrida no T6MM, a PAD, o índice cardíaco e o BNP/NT-proBNP são considerados fatores de prognóstico, devem ser avaliados regularmente e determinar a intensidade da terapêutica5.
No caso apresentado, os sintomas iniciaram-se 5 meses antes do internamento hospitalar e tiveram uma evolução rapidamente progressiva. Na fase inicial, o facto de ser columbófilo, conduziu a um diagnóstico apressado e incorreto, porque sem critérios, de alveolite de hipersensibilidade, o que atrasou a investigação e o diagnóstico da doença. Apesar da síndrome de apneia obstrutiva do sono poder ser uma causa de HP, não deve ser considerada, neste caso, dada a gravidade da HP e a evolução desfavorável apesar da instituição de VNI/CPAP. O CCD confirmou a presença de hipertensão pulmonar pré-capilar e o estudo etiológico permitiu excluir outras causas mais frequentes de HP. O doente não apresentava história de consumo de drogas ou tóxicos associadas à HAP, nem história familiar de HP, pelo que se pode admitir com grande segurança o diagnóstico de HAPi. À data do diagnóstico, o doente apresentava vários critérios de mau prognóstico pelo que tinha indicação para terapêutica combinada tripla. Nos anos mais recentes está cada vez mais preconizada a terapêutica combinada ad inicio, principalmente nos doentes que apresentam critérios de gravidade, como no caso apresentado1,8. Apesar da evolução clínica favorável com a terapêutica inicialmente instituída, não foi conseguida a normalização dos marcadores de prognóstico pelo que se substituiu o prostanóide inalado, pelo prostanóide endovenoso. Ao fim de 4 anos de seguimento, o doente mantém-se estável, autónomo nas suas atividades da vida diária, capaz de realizar exercício físico de intensidade moderada e com boa qualidade de vida.
O caso apresentado documenta a dificuldade e complexidade do diagnóstico da HAPi e a importância da referenciação precoce para um centro especializado, que no nosso país estão oficializados, no tratamento e seguimento destes doentes, bem como os bons resultados conseguidos com os tratamentos atualmente disponíveis que mudam radicalmente a funcionalidade, qualidade de vida e prognóstico desta doença.
Quadro I
Resumo da evolução dos marcadores de prognóstico da doença e medicação
| DATA | CLASSE NYHA | T6MM | Pro-BNP | TERAPÊUTICA ESPECÍFICA |
| | | (m) | (pg/mL) | |
| AGOSTO 2012 | IV | - | 1489 | Bos. + Ilop in. + sild. |
| OUTUBRO 2012 | II | 525 | 662,3 | Bos. + Ilop in. + sild. |
| FEVEREIRO 2013 | II | 549 | 1027 | Bos. + Ilop in. + sild. |
| FEVEREIRO 2014 | III | 525 | 1425 | Bos. + epo ev + sild. |
| AGOSTO 2014 | II | 592 | 994,7 | Bos. + epo ev + tad. |
| OUTUBRO 2015 | II | 600 | 731,6 | Mac. + epo ev + tad. |
| JULHO 2016 | II | 562 | 847,1 | Mac. + epo ev + tad. |
T6MM: teste de 6 minutos de marcha; Bos.: bosentano; Ilop. In: iloprost inalado; sild.: sildenafilo; epo ev: epoprostenol endovenoso; mac.: macitentano; tad.: tadalafil
Figura I
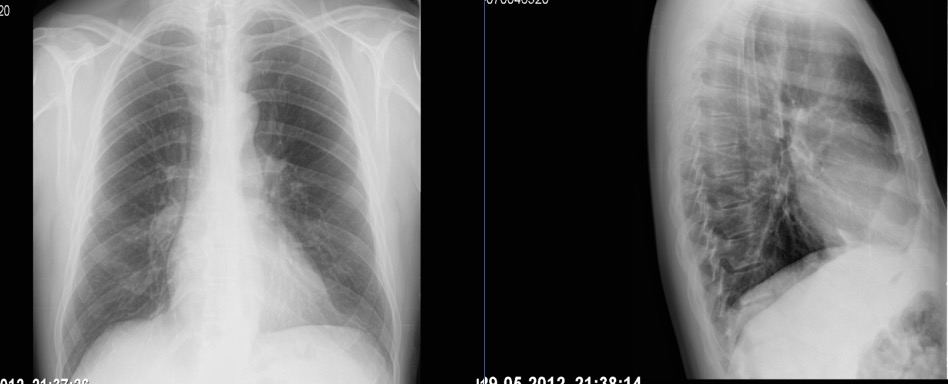
Telerradiografia de tórax: engurgitamento hilar bilateral; ICT >0,5 e alargamento do bordo direito cardíaco.
Figura II
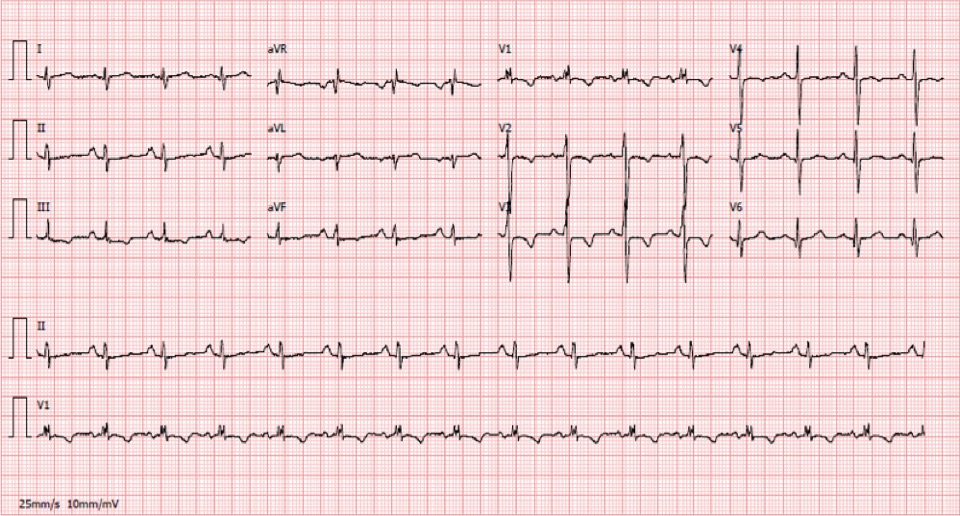
ECG: Ritmo sinusal, frequência cardíaca 70bpm; onda p pulmonar e bloqueio incompleto de ramo direito.
BIBLIOGRAFIA
1. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J, 2015.
2. Vakilian F, Attaran D, Shegofre M, Lari S, Chare S. Assessment of thyroid function in idiopathic pulmonary hypertension. Res Cardiovasc Med 2016 May; 5(2): e29361.
3. Bazan I, Fares W. Pulmonary hypertension: diagnostic and therapeutic challenges. Ther Clin Risk Manag 2015 Aug 17;11:1221-33.
4. Montani D, Gunther S, Dorfmuller P, Perros F, Girerd B et al. Orphanet J Rare Dis 2013 Jul 6;8:97.
5. Nickel N, Golpon H, Greer M, Knudsen L, Olsson K et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2012; 39: 589596.
6. Kiuchi M, Andrea B, Silva G, Coelho S, Paz L et al. Pulmonary artery ablation to treat pulmonary arterial hypertension: a case report. J Med Case Rep 2015 Dec 16;9:284.
7. Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani L, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J 2009(30): 394403.
8. Galiè N, Barbera JÁ, Frost AE, Ghofrani HÁ, Hoeper M, McLaughlin V, Peacock A Simonneau G, Vachiery JL, Grunig E, Oudiz RJ, Vonk-Noordegraaf A, White J, Blair C, Gilles H, Miller KL, Harris JHN, Langley J, Rubin LJ, for the AMBITION Investigators. N Engl J Med 2015; 373:834-844.

