INTRODUCTION:
A pancerebellar syndrome of subacute progression is highly suggestive of a paraneoplastic cerebellar degeneration (PCD).1 It is an uncommon paraneoplastic syndrome of the central nervous system (CNS) which usually develops within three months, has severity of at least 3 on the Rankin scale with no evidence of cerebellar atrophy.2 It is characterized by gait ataxia progressing to truncal and limb ataxia with dysarthria, nystagmus and diplopia.3
Typically it occurs in patients with ovarian, breast and lung cancer (especially small-cell lung-cancer) or Hodgkins lymphoma.4 PCD usually precedes the diagnosis of cancer and should elicit an investigation to find an underlying neoplasm. PCD may herald recurrence of a previous tumour or a second tumour.5
It is thought to be caused by antibodies synthesized within the brain by specific B cells that have crossed the blood-brain barrier6. Onconeural antibodies are thought to lead to a T-cell mediated destruction of Purkinje cells in the cerebellum.7 Only anti-Yo (or Purkinje cell antibody type 1 or PCA-1) and anti-Tr are predominantly associated with cerebellar dysfunction; others often associate with other areas of the CNS. Pathologic examination shows Purkinje cells degeneration along with inflammation in the cerebellar cortex, deep cerebellar nuclei and inferior olivary nuclei.8 An acute stage inflammation is thought to precede irreversible neuronal damage.9
Immediate treatment of the underlying tumour seems to be the best option but symptoms often remain.6,9 Patients with pronounced neuronal loss may respond to precocious immunotherapy: plasmapheresis, steroids, immunoglobulins, azathioprine, cyclophosphamide, rituximab (individually or in combination).10
Non-small cell lung carcinoma (NSCLC) accounts for 85% of all lung cancers. At the time of diagnosis the majority of patients have either locally advanced or metastatic disease.11
THE CASE:
We present a 76-year-old independent woman with hypertension and dyslipidemia. Her chronic medication was irrelevant. No hereditary diseases or smoking habits were reported.
At the age of 68 years old a 3-months pancerebellar syndrome forced her to move to her sons house. She presented left predominance gait ataxia, difficulties with high-precision manual tasks, 4 limb dysmetria and slight dysarthria. There was no toxic exposition nor new drugs prescribed. Her laboratory results are in Table 1 and 2.
Her electroencephalogram, head computed tomography (CT), brain magnetic resonance imaging (MRI) were normal. The thoracic and abdominopelvic CT, pelvic ecography, mammography and colonoscopy revealed no relevant findings. The positron emission tomography (PET) showed hyperfixation in her right posterior inferior lung. An extemporaneous biopsy revealed a bronchioloalveolar adenocarcinoma. The mass was removed through partial lobectomy which confirmed the diagnosis. The TNM classification was T1N0M0 and no further treatments were offered.
After surgery she could walk without help and regained full autonomy. Six months later new-onset walk unsteadiness and worsening dysarthria forced her to return to her son´s house. New PET and thoracic abdominal pelvic CT scan were performed and no evidence of cancer was found. Her brain CT showed cerebellar atrophy. After two cycles of Human Immunoglobulins with no improvement, treatment with methylprednisolone and cyclophosphamide (daily 1 g for 3 days) for 3 months was tried with partial response. However she could not live alone again. Clinical stability was achieved for 2 months but her dysarthria and ataxia began worsening. There was no clear response to rituximab (one cycle of 375 mg/m2 for 3 months, after approval by the Hospital Ethics Committee) nor to a new cycle of methylprednisone (1 g). Her dysarthria improved significantly and her gait had a slight recovery after plasmapheresis (3 days per week during a month and then weekly for 2 months) and a cycle of rituximab with plasmapheresis. She was also under motor rehabilitation.
After one year, a new relapse determined intensive plasmapheresis (daily for 5 days) and she began mophetil mycophenolate (slowing increasing doses until 1,5 gr daily) with clinical stability until now (Fig. 1)
DISCUSSION:
This 76 years-old woman has developed NSCLC with PCD. There was mild protein elevation and no CSF pleocytosis. A previous work showed CSF pleocytosis and protein normalization after 3 months of neurological symptom onset.12 In our case, CSF analysis was made 3 months after the complaints. However, a monoclonal band in CSF was found which could be related with intrathecal inflammation. No onconeural antibodies were detected and no significant cerebellar lesions were initially found but diffuse cerebellar atrophy eventually appeared as expected. Cases of PCD in patients with NSCLC without detectable onconeural antibodies have been described but they were included in larger series of different tumours or have been reported as single observations.13 Studies with larger series are needed.
In our case we believe a potential reversible immune-mediated answer was achieved under cyclophosphamide, combination of plasmapheresis and rituximab and with mycophenolate mophetil. Immunoglobulins and methylprednisolone were not effective. The relative roles of humoral and cell mediated immunity in paraneoplastic neurologic disorders are unresolved. For instance, in Lambert-Eaton myasthenic syndrome or myasthenia gravis antibodies have a role in the pathogenesis as the target antigens are cell-surface receptors.14 Therefore their response to plasma exchange or intravenous immune globulin is good. Vesicle-associated proteins in the presynaptic nerve terminal are another antibody target. In the stiff-man syndrome the antibody is taken up in the nerve terminal and bind soluble proteins in the nerve terminus. Neuron-specific RNA-binding proteins (nuclear molecules) and neuronal signal transduction proteins (cytosolic proteins) are two classes of intracellular proteins which T-cells may recognize as an MHC-peptide complex eliciting a cytotoxic attack.14
In this case, response to rituximab may be the consequence of depleting the autoreactive B-Cell compartment after CD-20 positive B-Cell depletion. Response to cyclophosphamide, cortisone and mycophenolate mophetil may be due to concomitant suppression of T-cell response. Poor outcome with human immunoglobulin may result of low susceptibility of intrathecal inflammation to its humoral systemic effects.15 Later use of plasma exchange intended to clear potential autoantibodies in serum but probably systemic antibodies were not an important pathological feature. We speculate that a cell-mediated immune response thus not humoral - is the core mechanism and there can still be a subacute inflammation causing symptoms and neuronal damage.
Rapid diagnosis, aggressive and combined treatment, even in the absence of immunological markers are essential to prevent ongoing neurological injury.
She is still alive almost 9 years after her diagnosis. The european 5-year overall survival rate is 73% (stage Ia) but it falls to less than 50% in advanced stages.11 Early recognition of paraneoplastic cerebellar degeneration can improve prognosis by treating neoplasms at an early stage.
Figura I
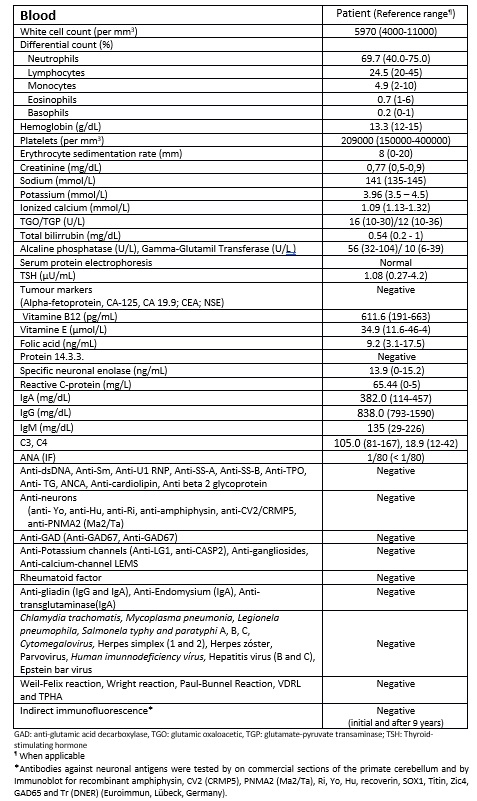
Table 1 - Blood testing
Figura II
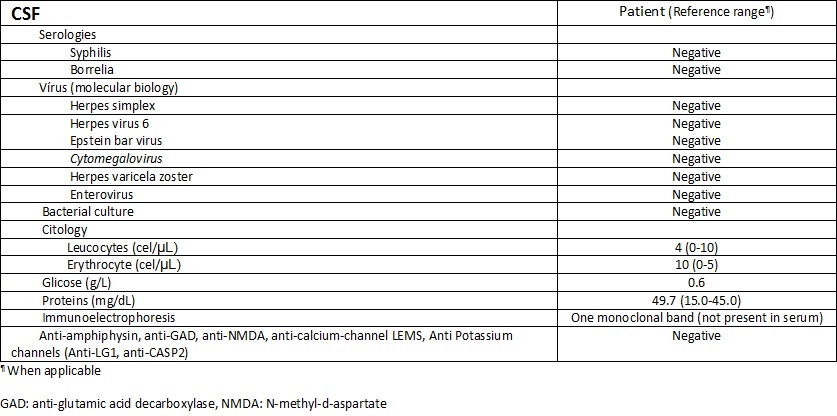
Table 2 - CSF testing
Figura III
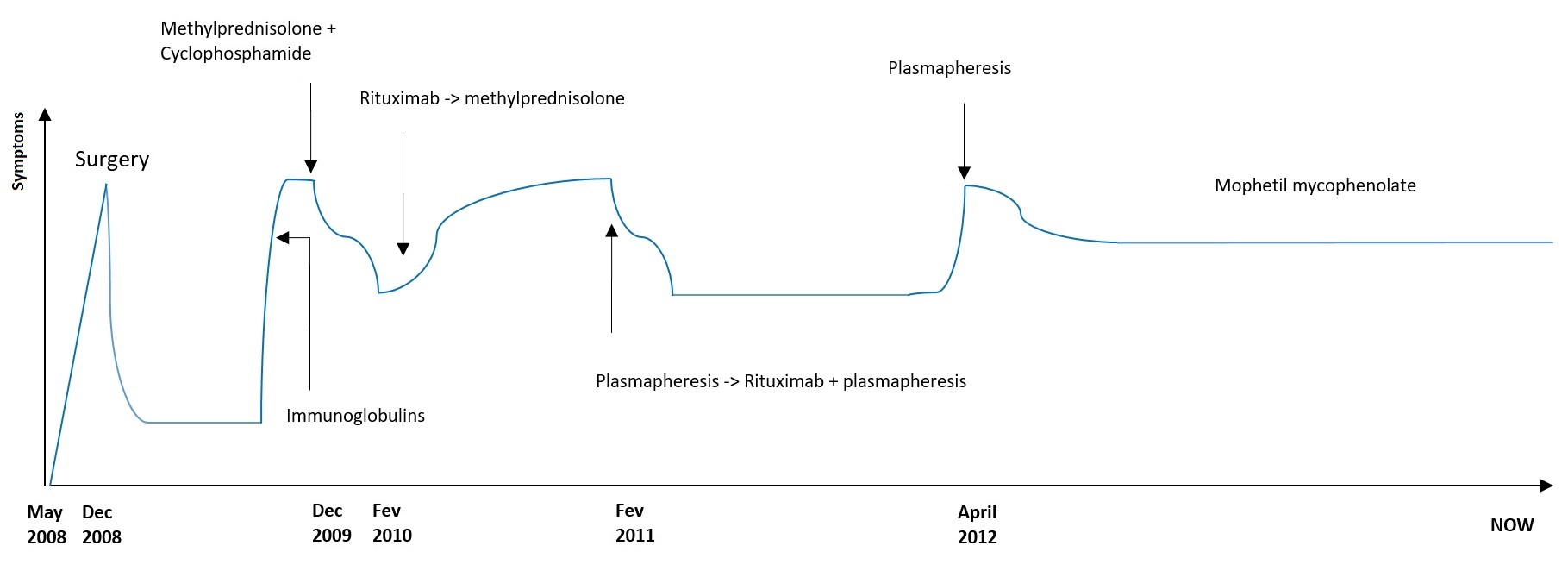
Clinical evolution and treatments
BIBLIOGRAFIA
1 Honnorat J et al. Paraneoplastic neurological syndromes. Orphanet J Rare Dis 2007; 2: 22;
2 Graus F et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 2004; 75: 1135-1140
3 C. A. Vedeler et al, Management of paraneoplastic neurological syndromes: report of an EFNS Task Force; European Journal of Neurology, 2006, vol. 13, no. 7, pp. 682690.
4 SL, Posner JB. Autoimmune response of patients with paraneoplastic cerebellar degeneration to a Purkinje cell cytoplasmic protein antigen. Ann Neurol, 1985; 18:592-600
5 de Beukelaar, J, Managing Paraneoplastic Neurological Disorders. The Oncologist 2006; 11: 292305
6 Darnell R B et al. Paraneoplastic Syndromes Involving the Nervous System. N Engl J Med 2003; 349:1543-54
7 Jaeckle KA et al. Autoimmune response of patients with paraneoplastic cerebellar degeneration to a Purkinje cell cytoplasmic protein antigen. Ann Neurol 1985;18:592-600.
8 Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol 2008; 7:327.
9 Storstein A et al. Treatment of Paraneoplastic Neurological Syndromes. Adv Clin Chem. 2007; 44:143-85.
10 Dalmau, J et al. Paraneoplastic cerebellar degeneration, Uptodate. Retrieved 26/06/2016 from http://www.uptodate.com/contents/paraneoplastic-cerebellar-degeneration
11 Vansteenkiste, J et al. Early and locally advanced non-small cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow up. Annals of Oncology 24 (Supplement 6): vi89vi98, 2013
12 Psimaras D. et al. Euronetwork PNS. Cerebrospinal fluid study in paraneoplastic syndromes, J Neurol Neurosurg Psychiatry 2010;81:425.
13 Sabater L et al, Protein kinase Cc autoimmunity in paraneoplastic cerebellar degeneration and non-small-cell lung cancer, J Neurol Neurosurg Psychiatry, 2006;77:13591362.
14 Darnell RB. Onconeural antigens and the paraneoplastic neurologic disorders: at the intersection of cancer, immunity, and the brain. Proc Natl Acad Sci U S A 1996; 93(10):45294536.
15 E Wiles C M et al. Intravenous immunoglobulin in neurological disease: a specialist review. J Neurol Neurosurg Psychiatry 2002;72:440448

