

Introdução
A Poliangeíte Microscópica (PAM) é uma vasculite necrotizante pauci-imune não granulomatosa sistémica que afeta os pequenos vasos e caracteriza-se por anticorpo anti-neutrofílico citoplasmático perinuclear (pANCA) positivo1. Podem ocorrer manifestações multissistémicas, incluindo sintomas constitucionais. Os sintomas prodrómicos podem durar semanas a meses, sem evidência de envolvimento específico de orgão, dificultando o diagnóstico desta patologia. O diagnóstico é baseado no exame físico dos orgãos envolvidos, avaliação analítica e radiografia ou tomografia computorizada (TC) de tórax e é confirmado através de biopsia tecidular1. O tratamento consiste em terapêutica imunossupressora.
Caso Clínico
Doente do sexo feminino, 62 anos, natural de Lisboa, reformada (doméstica), que recorreu ao Serviço de Urgência por síndrome gripal, febre e tosse produtiva com 10 dias de evolução. Negava toracalgia, dispneia, perda de peso, anorexia ou outros sintomas acompanhantes.
Apresentava antecedentes pessoais de neoplasia da mama com status pós-mastectomia e hormonoterapia com tamoxifeno que cumpriu durante 6 anos, hipertensão arterial, esofagite de refluxo e safenectomia esquerda. Negava hábitos tabágicos e alcoólicos. Negava alergias. Estava medicada com Omeprazol 20 mg, Estazolam 2 mg e multivitamínico.
Na observação destacava-se pressão arterial 130/75 mmHg, frequência cardíaca 70 bpm, frequência respiratória 22 cpm, auscultação pulmonar com murmúrio vesicular diminuído nas bases e fervores sub-crepitantes bibasais.
Analiticamente com hemoglobina 8,2 g/dL, creatinina 4,20 mg/dL, ureia 119 mg/dL, VS 111 mm, PCR 3,16 g/dL, sedimento urinário inocente, sem proteinúria na urina de 24 horas.
Realizou radiografia de tórax que mostrou condensação heterogénea nos dois terços inferiores do hemitórax direito (Fig. 1) e ecografia renal com perda de diferenciação parenquimo-sinusal e com aumento difuso da ecogenicidade do parênquima, alterações compatíveis com nefropatia médica.
Ficou internada e iniciou antibioterapia empírica com ceftriaxone por suspeita de pneumonia, não se tendo registado melhoria do quadro. As hemoculturas, urocultura e exame microbiológico da expetoração com pesquisa de Mycobacterium tuberculosis foram negativos, bom como as serologias virais (HIV, HCV e HBV) e bacterianas (Mycoplasma pneumoniae, Legionella pneumophila e Chlamydia pneumoniae).
Durante o internamento iniciou quadro de hemoptises pelo que foi efetuada TC de tórax que revelou alterações do parênquima pulmonar traduzidas por múltiplas áreas de densificação, associadas a espessamento intralobular, com padrão essencialmente alveolar (Fig. 2). Neste contexto, foi pedida broncofibroscopia que identificou hemorragia proveniente do lobo inferior direito. A biopsia brônquica demonstrou infiltrado inflamatório de tipo crónico, inespecífico e o lavado bronco-alveolar foi negativo para células neoplásicas e Pneumocystis jiroveci.
Após discussão com a Nefrologia, optou-se por biopsia renal onde se constatou glomerulonefrite necrotizante pauci-imune com crescentes (Fig. 3). O pANCA foi positivo, com restante estudo autoimune negativo, sem consumo de complemento e doseamento de crioglobulinas negativo. Perante estas alterações foi colocada a hipótese de PAM. Após oito dias de internamento, foi iniciada terapêutica com prednisolona 1 mg/kg/dia, ciclofosfamida 1,5 mg/kg/dia e darbepoietina 60 μg/semana.
Houve melhoria clínica, analítica e radiológica (Fig. 4 e 5) significativas, destacando-se, analiticamente à data de alta, hemoglobina 9,2 g/dL, creatinina 2,50 mg/dL e VS 13 mm.
Em ambulatório manteve seguimento conjunto com a Medicina Interna e a Nefrologia. Cumpriu 3 meses de ciclofosfamida e iniciou azatioprina 2 mg/kg/dia aos 4 meses, simultaneamente com desmame lento de prednisolona até 5 mg/dia. Ocorreu remissão clínica ao fim de um ano de tratamento. Nesta altura apresentava hemoglobina 13,3 g/dL, creatinina 1,60 mg/dL, VS 21 mm e pANCA negativo, tendo suspendido a terapêutica.
Dois anos após remissão, iniciou queixas oculares com quadro de uveíte, que motivou reintrodução de prednisolona e azatioprina. Foi ainda adicionado alopurinol.
Nesta sequência desenvolveu anemia que motivou novo internamento. O quadro foi interpretado como iatrogenia à medicação com azatioprina e alopurinol, fármacos que foram suspensos, com melhoria.
Foi doseada a tiopurina metiltransferase (TPMT) tendo sido normal, pelo que reiniciou azatioprina 1 mg/kg/dia, prednisolona 10 mg/dia e darbepoietina 40 μg/semana. Manteve-se sem alopurinol.
Permaneceu estável durante cerca de 1 ano, com posterior agravamento progressivo da função renal (creatinina 6,00 mg/dL) e indicação atual para hemodiálise.
Discussão
O caso clínico representa o quadro de um síndrome pulmão-rim, caracterizado por uma associação de hemorragia alveolar difusa e glomerulonefrite que é característico da PAM. Nesta patologia pode ainda ocorrer envolvimento cutâneo (púrpura palpável, petéquias, livedo reticularis, eritema palmar, nódulos cutâneos)2, neurológico (mononeurite múltipla, envolvimento do sistema nervoso central)3, gastrointestinal (hemorragia, ulceração, pancreatite)4 ou ocular (vasculite retiniana, esclerite)5 e os doentes geralmente apresentam sintomas constitucionais como febre, mal-estar geral, mialgias, artralgias, anorexia e perda de peso6. A tabela 1 retrata o diagnóstico diferencial a considerar nos síndromes pulmão-rim7.
A nível laboratorial pode ser detetada anemia normocítica normocrómica, leucocitose e/ou trombocitose, velocidade de sedimentação aumentada, creatinina aumentada, sedimento urinário ativo, com proteinúria ou hematúria e pANCA positivo. O pANCA é positivo em cerca de 80% dos casos de PAM, encontrando-se especialmente elevado em doentes com envolvimento renal isolado2. Devem ainda ser realizados exames laboratoriais de forma a excluir outros diagnósticos, nomeadamente anticorpos antinucleares (ANA), fatores do complemento (C3 e C4), crioglobulinas, serologias de vírus hepatotrópicos, HIV, testes de função hepática e hemoculturas.
Os exames imagiológicos do tórax podem mostrar opacificações bilaterais compatíveis com hemorragia alveolar ou espessamento das grandes vias aéreas, correspondente a infiltração por linfócitos e fibrose8.
Para confirmação do diagnóstico é necessária a realização de biopsia de tecido afetado, nomeadamente pele, pulmão, rim, músculo ou nervo sural. A histologia pode revelar uma arterite necrotizante que poupa os grandes vasos e pode estar presente necrose fibrinoide ou infiltração por neutrófilos fragmentados na parede vascular, denominada por vasculite leucocitoclástica9. A glomerulonefrite é caracterizada por necrose focal, formação de crescentes e ausência ou escassez de depósitos de imunoglobulinas10. A biopsia renal foi essencial para estabelecer o diagnóstico no caso clínico apresentado.
O tratamento inicial (fase de remissão) consiste em ciclofosfamida 1,5-2 mg/kg/dia e glucocorticoides - pulso de metilprednisolona seguido de prednisolona 1mg/kg/dia. Não está indicada monoterapia com glucocorticoides, por se associar a baixa taxa de remissão11. Na fase de manutenção a ciclofosfamida deve ser substituída por metotrexato ou azatioprina devido à sua toxicidade. Tem sido proposta terapêutica com rituximab que se mostrou não inferior à ciclofosfamida na indução da remissão da vasculite associada a ANCA12 e foi bem tolerada no tratamento de manutenção. Não existem ainda estudos conclusivos relativamente à prevenção de recaídas13.
A doente iniciou a terapêutica recomendada 18 dias após o início dos sintomas, com melhoria do quadro clínico, tendo, no entanto, havido recaída. Realizou 3 meses de ciclofosfamida o que poderia eventualmente ter contribuído para um aumento do risco de recaída, tal como demonstrado num estudo por Guillevin L, no qual se registou superioridade na prevenção da recaída na terapêutica com 12 meses de ciclofosfamida relativamente à terapêutica com 6 meses14. Por outro lado, foram realizados esforços para um diagnóstico rápido, de modo a que a terapêutica fosse iniciada atempadamente, o que constitui igualmente um fator de prevenção de recaída15.
Como terapêutica de manutenção, a doente manteve imunossupressão com azatioprina, escolhida em detrimento do metotrexato devido ao contexto de insuficiência renal. Esta consiste num derivado imidazólico da 6-mercaptopurina que atua como antimetabolito, inibindo a síntese de ácidos nucleicos. Na evolução da sua doença ocorreu intoxicação por este fármaco, tendo sido realizado o doseamento da tiopurina-metiltransferase (TPMT) que foi normal. Este episódio foi então interpretado como interação medicamentosa com alopurinol, o qual aumenta a intoxicação pela azatioprina por inibição da enzima xantina oxidase e, desta forma, de uma das vias de metabolização da azatioprina. A azatioprina é metabolizada no fígado e nos eritrócitos, tendo a TPMT uma ação preponderante na sua metabolização.
Apesar da terapêutica de manutenção, acabou por ocorrer agravamento da função renal com atual indicação para diálise. A evolução da PAM para insuficiência renal crónica terminal, permanece uma das principais complicações desta patologia, ocorrendo em cerca de 30% dos casos15.
Apresenta-se assim o caso de uma vasculite sistémica ANCA positiva com progressão para insuficiência renal terminal, evolução que consiste num dos grandes desafios terapêuticos desta patologia.
Quadro I
Diagnóstico diferencial de Síndromes pulmão-rim (Adaptado de Papiris SA et al. 2007)
| Diagnóstico Diferencial de Síndrome Pulmão-Rim |
| Síndrome pulmão-rim associado a anticorpo anti-GBM |
| Síndrome de Goodpasture |
| Síndrome pulmão-rim nas vasculites sistémicas ANCA-positivas |
| Granulomatose de Wegener; Poliangeíte microscópica; Síndrome Churg-Strauss; outras vasculites |
| Síndrome pulmão-rim nas vasculites sistémicas ANCA-negativas |
| Púrpura de Henoch-Schönlein; Crioglobulinemia mista essencial; Doença de Behçet; Nefropatia por IgA |
| Síndrome pulmão-rim ANCA-positivo sem vasculite sistémica: síndrome pulmão-rim idiopático |
| Glomerulonefrite necrótica pauci-imune e capilarite pulmonar |
| Síndrome pulmão-rim na vasculite ANCA-positivo associada a fármacos |
| Propiltiouracilo, penicilamina, hidralazina, alopurinol, sulfasalazina |
| Síndrome pulmão-rim em doentes com anti-GBM e ANCA positivos |
| Síndrome pulmão-rim associado a doenças autoimunes (mediadas por imunocomplexos ou ANCA) |
| Lúpus eritematoso sistémico; Esclerodermia; Polimiosite; Artrite reumatóide; Doença mista do colagénio vascular |
| Síndrome pulmão-rim associado a microangiopatia trombótica |
| Síndrome anti-fosfolípido; Púrpura trombocitopénica trombótica; Infecção; Neoplasia |
| Glomerulonefrite pauci-imune idiopática complicada com hemorragia alveolar difusa |
ANCA - anticorpo anti-neutrofílico citoplasmático; GBM - membrana basal glomerular
Figura I
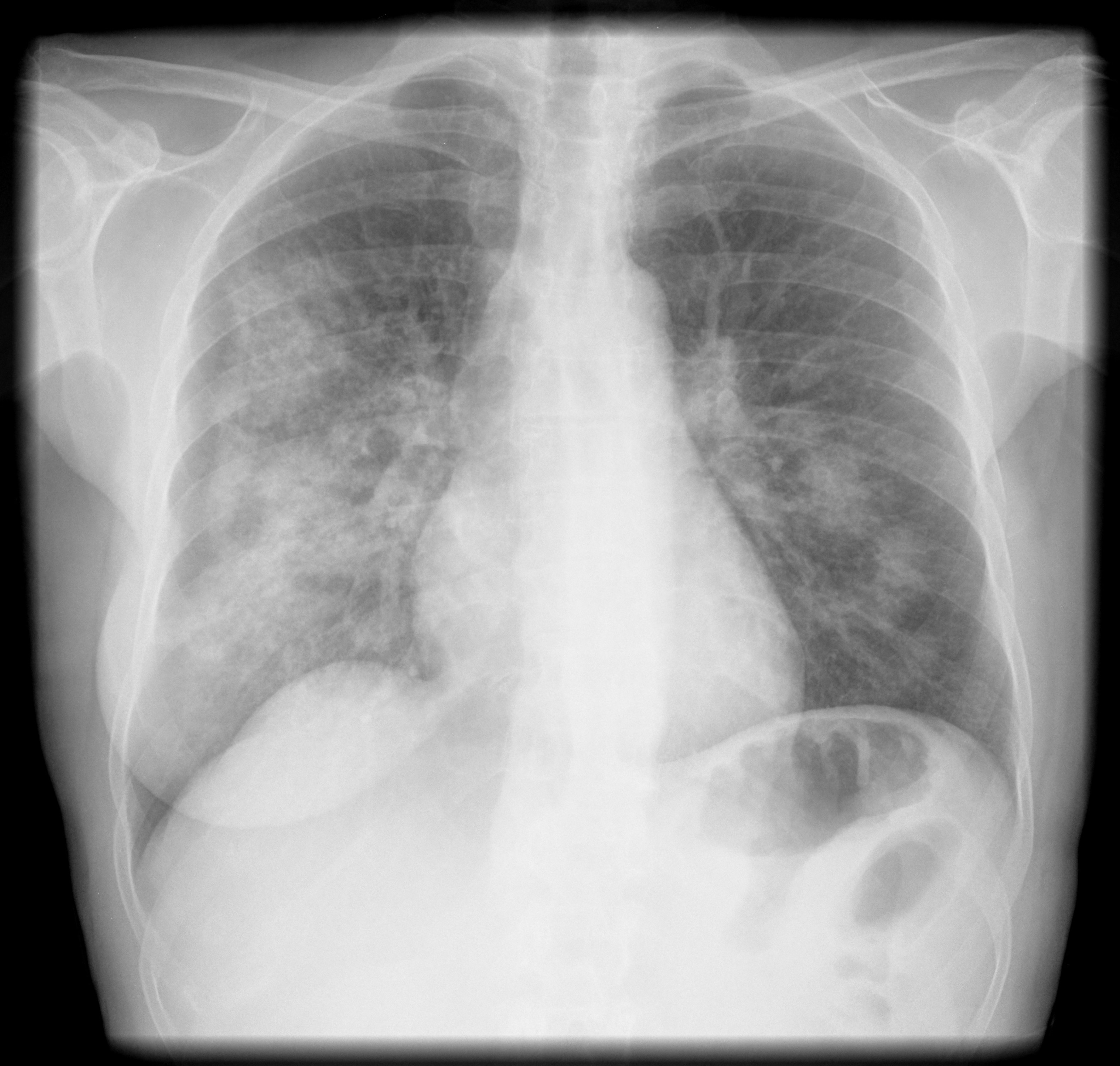
Radiografia de tórax com condensação heterogénea nos dois terços inferiores do hemitórax direito
Figura II
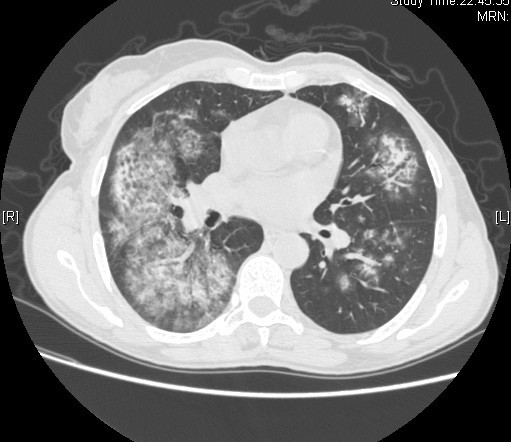
TC de tórax com múltiplas áreas de densificação, associadas a espessamento intralobular, com padrão essencialmente alveolar
Figura III
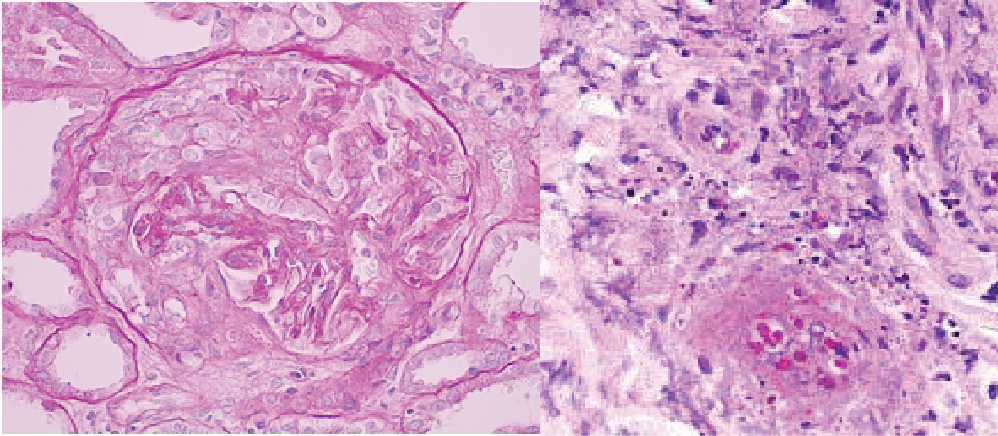
Biopsia renal com glomerulonefrite necrotizante pauci-imune com crescentes
Figura IV
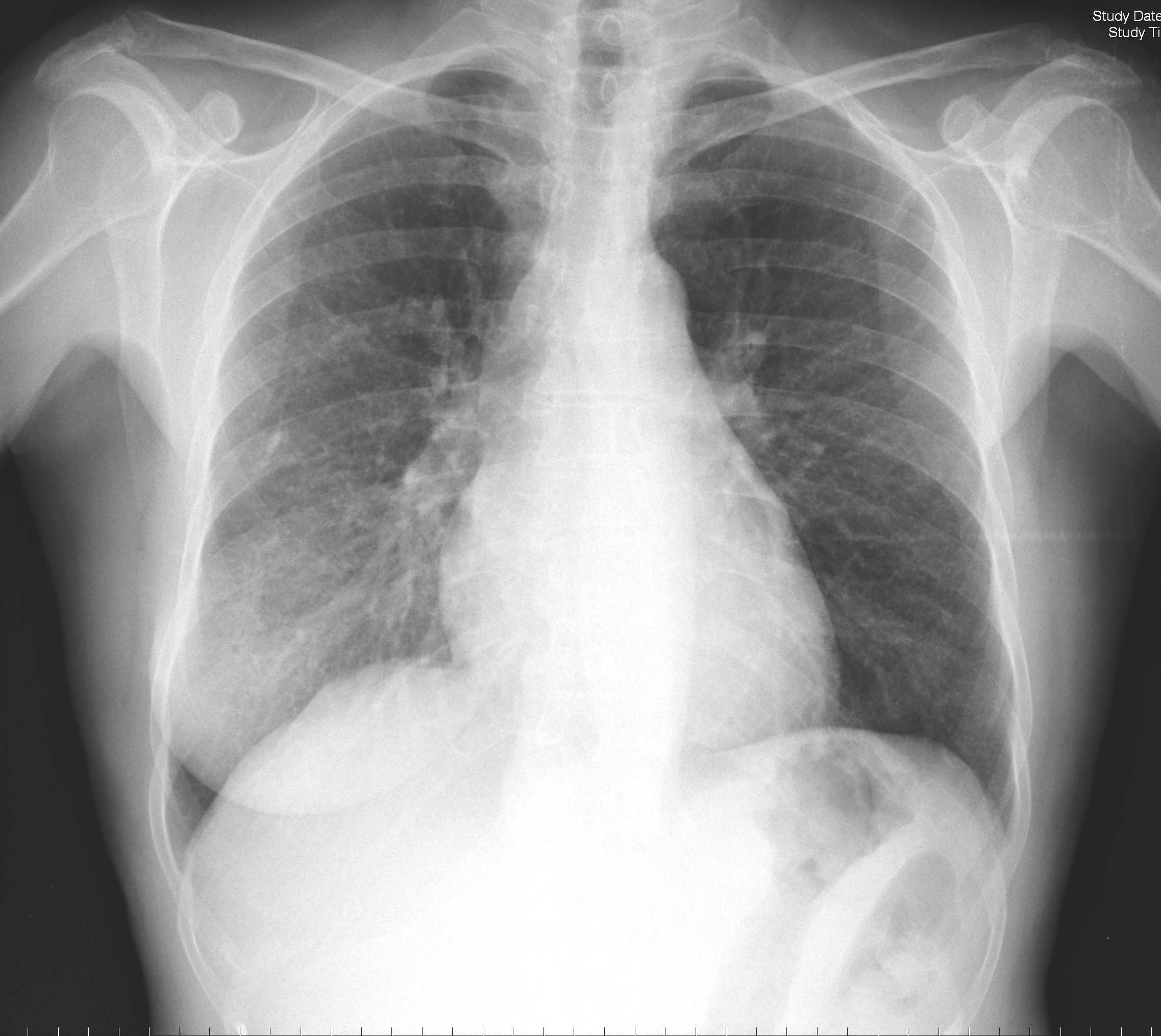
Radiografia tórax à data de alta com resolução da condensação
Figura V
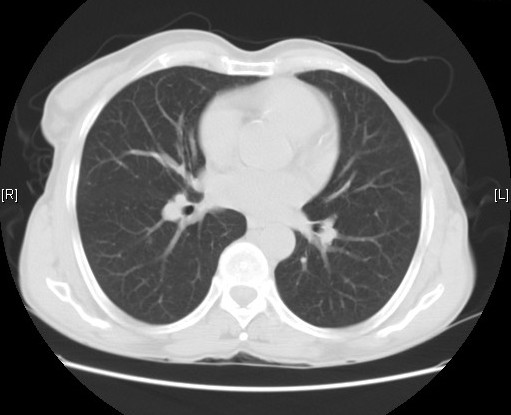
TC tórax à data de alta com resolução das alterações parenquimatosas
Figura VI
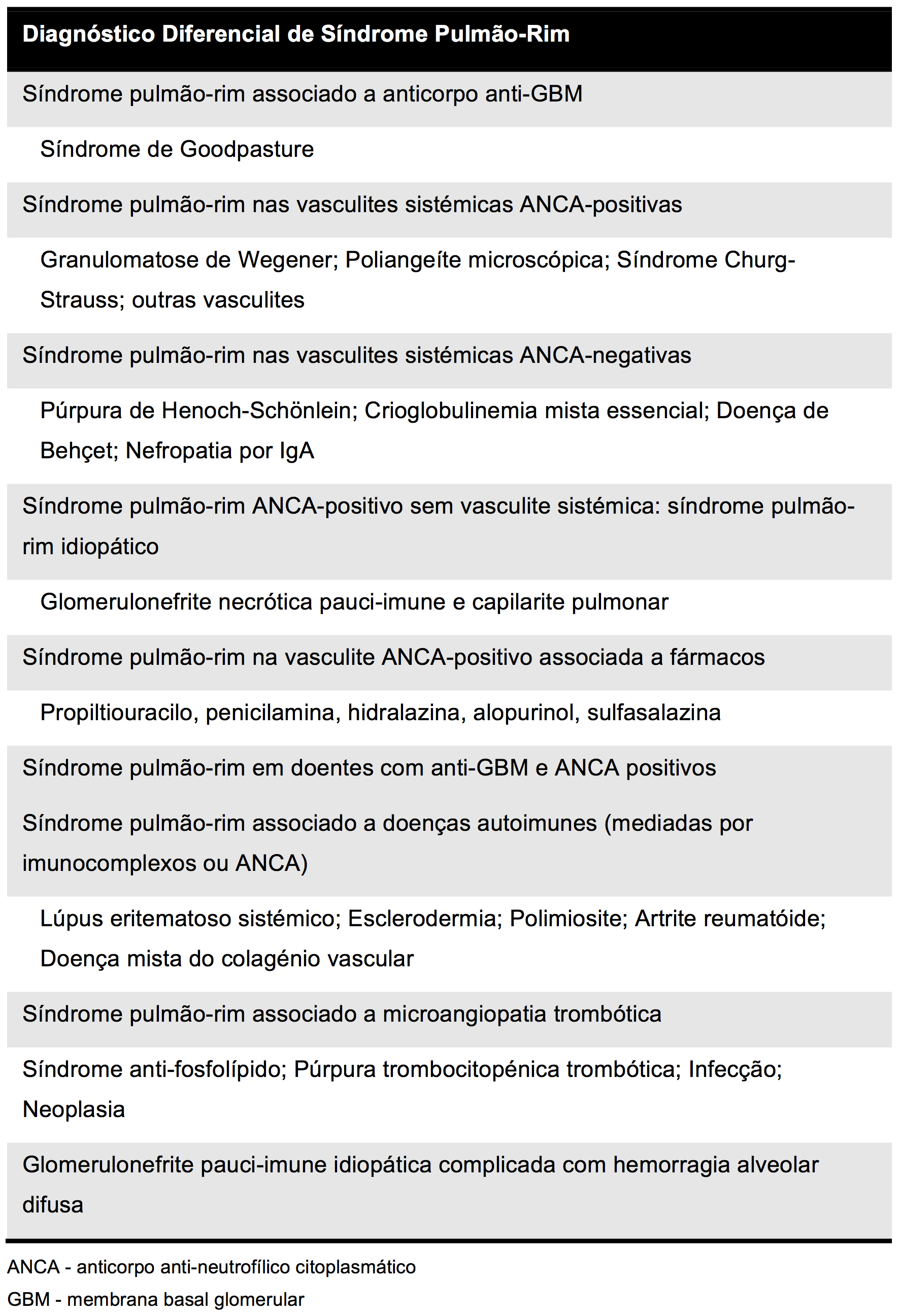
Imagem referente à tabela 1 para melhor percepção do efeito pretendido
BIBLIOGRAFIA
1. Jennette JC, Falk RJ, Andrassy K, Bacon P a, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37(2):18792.
2. Carlson J. Cutaneous vasculitis. In: Dermatopathology: A Volume in the Foundations in Diagnostic Pathology Series. USA: Saunders Elsevier; 2010. p. 184208.
3. Sassi S Ben, Ghorbel I Ben, Mizouni H, Houman MH, Hentati F. Microscopic polyangiitis presenting with peripheral and central neurological manifestations. Neurol Sci. 2011 Aug;32(4):7279.
4. Pagnoux C, Mahr A, Cohen P, Guillevin L. Presentation and Outcome of Gastrointestinal Involvement in Systemic Necrotizing Vasculitides. Medicine (Baltimore). 2005;84(2):11528.
5. McCluskey P, Powell RJ. The eye in systemic inflammatory diseases. Lancet. 2004;364:212533.
6. Agard C, Mouthon L, Mahr A, Guillevin L. Microscopic polyangiitis and polyarteritis nodosa: how and when do they start? Arthritis Rheum. 2003;49(5):70915.
7. Papiris S a, Manali ED, Kalomenidis I, Kapotsis GE, Karakatsani A, Roussos C. Bench-to-bedside review: pulmonary-renal syndromes--an update for the intensivist. Crit Care. BioMed Central. 2007;11(3):213.
8. Collins CE, Quismorio FP. Pulmonary involvement in microscopic polyangiitis. Curr Opin inPulmonary Med. 2005;11:44751.
9. Kawakami T, Soma Y, Saito C, Ogawa H, Nagahuchi Y, Okazaki T, et al. Cutaneous manifestations in patients with microscopic polyangiitis: Two case reports and a minireview. Acta Derm Venereol. 2006;86(2):1447.
10. Haas M, Eustace JA. Immune complex deposits in ANCA-associated crescentic glomerulonephritis: A study of 126 cases. Kidney Int. 2004;65(6):214552.
11. Nachman PH, Hogan SL, Jennette JC, Falk RJ. Treatment response and relapse in antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol. 1996;7:339.
12. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus Cyclophosphamide for ANCA-Associated Vasculitis. N Engl J Med. 2010;363(3):22132.
13. Roubaud-Baudron C, Pagnoux C, Méaux-Ruault N, Grasland A, Zoulim A, Guen J LE, et al. Rituximab Maintenance Therapy for Granulomatosis with Polyangiitis and Microscopic Polyangiitis. J Rheumatol. 2012 Jan 1;39(1):12530.
14. Guillevin L, Cohen P, Mahr A, Arène J-P, Mouthon L, Puéchal X, et al. Treatment of polyarteritis nodosa and microscopic polyangiitis with poor prognosis factors: a prospective trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in sixty-five patients. J Clin Rheumatol. 2014;20(4):17982.
15. Corral-Gudino L, Borao-Cengotita-Bengoa M, Del Pino-Montes J, Lerma-Má Rquez JL. Overall survival, renal survival and relapse in patients with microscopic polyangiitis: a systematic review of current evidence. Rheumatology. 2011;50(8):141423.