

Introdução:
As miopatias autoimunes são um grupo raro e heterogéneo de patologias que afetam duas vezes mais o sexo feminino.1,3-5 Apresentam uma incidência de 4 em 100 000 pessoas por ano e prevalência de 15 a 32 casos em 100 000 pessoas.1 Tipicamente apresentam perda de força muscular proximal, simétrica e progressiva em semanas a meses.1-3 No entanto, cada subtipo de miopatia autoimune está associado a características clínicas, histopatológicas e patofisiológicas distintas.1-3,7,9
A síndrome anti-sintetase é um subtipo de miopatia autoimune que pela sua raridade apresenta prevalência desconhecida. Além disso, os estudos são escassos.4 Esta síndrome inclui duas ou mais das seguintes características: miosite; patologia intersticial pulmonar; artrite; fenómeno de Raynaud; febre; ou lesões hiperqueratóticas radiais e palmares dos dedos, denominadas mãos de mecânico.1,3-5 Também é caraterizado pela presença de auto-anticorpos contra as aminoacil-tRNA sintetases.1-5
Caso Clínico:
Este trabalho relata um caso clínico de um homem de 63 anos com antecedentes de fenómeno de Raynaud com vários anos de evolução, hipertensão arterial, tabagismo e hábitos alcoólicos (cerca de 40g/dia).
Um mês antes da admissão iniciou um quadro anorexia, perda ponderal que não sabia quantificar, enfartamento pós-prandial, vómitos, disfagia, dejeções diarreicas, dispneia para pequenos esforços e diminuição da força muscular de predomínio proximal e simétrica.
Recorreu ao Serviço de Urgência por febre de predomínio noturno associada a dor e rubor do flanco esquerdo com 3 dias de evolução.
À admissão apresentava-se febril (38,4ºC), taquicárdico (FC 110 bpm), eupneico com SpO2 (FiO2 21%) 95% e com sinais inflamatórios no flanco esquerdo. Analiticamente apresentava desidrogenase do lactato (DHL) 519 U/L, creatinafosfoquinase (CK) 1059 U/L, sem leucocitose ou neutrofilia e proteína C reativa 5,77 mg/dL. A gasimetria arterial (fiO2 21%) apresentava: alcalemia (pH 7,466) com alcalose respiratória (pCO2 29,1 mmHg), HCO3- 22,4 mmol/L, hipoxemia (pO2 69,4 mmHg), e lactatos 1,7 mmol/L com hiato aniónico normal. A teleradiografia de tórax apresentava subida da hemicúpula esquerda. A ecografia abdominal demonstrou que a tumefação do flanco esquerdo no exame objetivo não apresentava tradução ecográfica, hepatomegalia com aumento difuso e homogéneo da sua ecogenecidade. Iniciou antibioterapia empírica com amoxicilina/ácido clavulânico e clindamicina e foi internado no serviço de Medicina Interna com o diagnóstico de celulite da parede abdominal e para estudo da síndrome constitucional.
Durante o internamento efetuou: endoscopia digestiva alta que demonstrou candidose esofágica, tendo iniciado fluconazol; e a endoscopia digestiva baixa apresentou pólipos sésseis.
Por manifestar telangiectasias da face, diminuição da força muscular proximal, hiperpigmentação em V no tórax, lesões vinosas nas articulações metacarpofalangicas da mão direita (Pápulas de Gottron), por persistência da CK elevada e aldolase elevada (31,5 U/L) foi pedido estudo imunológico. Este apresentava Velocidade de sedimentação 78 mm/Hr; anticorpos anti-nucleares (ANAs) com título 1/1280 e padrão nucleolar e mosqueado, anticorpo anti-Jo1 e anti-Ro 52 positivos. A capilaroscopia apresentava megacapilares, tortuosos, desorganizados e microhemorragias, sem áreas de rarefação, sendo este padrão sugestivo de dermatomiosite. (Fig. 1 e 2)
Por insuficiência respiratória agravada com necessidade de ventilação mecânica invasiva efetuou Tomografia computorizada do tórax (TC tórax) que evidenciou áreas de consolidação parenquimatosa irregular e mal definida na região subpleural posterior dos lobos superiores e inferiores bilateralmente, associadas a vidro despolido, espessamento peri-broncovascular em algumas áreas com preservação de banda parenquimatosa subpleural, aspetos compatíveis com envolvimento pulmonar de patologia do tecido conjuntivo. (Fig. 3 e 4)
Durante o internamento apresentou padrão de citocolestase hepática persistente e após exclusão de outras etiologias foi demonstrado anti SLA/LP positivo forte. No entanto, foi recusada pelo paciente a realização de biopsia hepática.
Assim, foi efetuado o diagnóstico de Síndrome anti-sintetase com anti-Jo1 e anti-Ro 52 positivos, com Pneumonia intersticial não específica (NSIP) e hepatite autoimune provável.
Iniciou corticoterapia com pulsos de metilprednisolona 1g/dia, seguidos de prednisolona oral.
Efetuadas biópsias muscular do deltóide e cutânea que não foram conclusivas. A ausência de alterações pode dever-se a terem sido efetuadas após o início da corticoterapia.
Evidenciada progressiva melhoria clínica e imagiológica (Fig. 5 e 6) com possibilidade de alta com corticoterapia em desmame e encaminhamento para a consulta externa.
Em regime de ambulatório efetuou: ecocardiograma transtorácico que não demonstrou alterações de relevo; e provas de função respiratória que demonstraram alteração ventilatória restritiva ligeira com defeito de difusão de CO (DLCO).
O caso foi discutido em reunião multidisciplinar de doenças autoimunes, sendo decidido o início de terapêutica com imunoglobulinas. A escolha desta terapêutica deveu-se à melhoria do atingimento pulmonar da doença, não apresentando nessa altura indicação para terapêutica imunossupressora específica dirigida ao órgão, nomeadamente ciclofosfamida; ausência de resposta muscular aos corticoides; presença de provável hepatite autoimune; presença de tuberculose latente com toxicidade hepática e cutânea à isoniazida e rifampicina, o que limitou o uso de outros imunossupressores nomeadamente o rituximab. Após o primeiro ciclo de imunoglobulinas o doente apresentou melhoria da força muscular e analítica.
Discussão:
A síndrome anti-sintetase é um subtipo de miopatia autoimune que inclui duas ou mais das seguintes características: miosite, com aumento da CK, DHL e aldolase; patologia intersticial pulmonar; artrite; fenómeno de Raynaud; febre; ou lesões hiperqueratóticas radiais e palmares dos dedos, denominadas mãos de mecânico.1,3-5 Além disso, podem apresentar exantemas semelhantes aos dos doentes com dermatomiosite1, assim como as pápulas de Gottron, estas últimas presentes em 20% dos casos.5
Também é caracterizado pela presença de auto-anticorpos contra as aminoacil-tRNA sintetases, uma família de enzimas intra-citoplasmáticas que desempenham um papel vital na síntese proteica.1-6 Estes auto-anticorpos estão presentes em 20% dos doentes com miopatias auto-imunes1, sendo o anti-Jo1 o mais frequente.2,5,8,9 A fraqueza muscular, artrite e as mãos de mecânico são mais comuns na presença do anti-Jo1.2 A presença de anti-Jo1 está associada a pior prognóstico.3 A biopsia muscular de doentes com anti-Jo1 positivo apresentam um padrão específico de necrose perifascicular com deposição de sarcolema, que se diferencia da atrofia perifascicular dos doentes com dermatomiosite.1 No entanto, o exame histológico não é obrigatório para o diagnóstico.5 A presença de anti-Ro tem sido associada à fibrose pulmonar3 e à patologia intersticial pulmonar, assim como ao atingimento articular e muscular mais grave e à maior probabilidade de neoplasia.5
A prevalência de patologia intersticial pulmonar na síndrome anti-sintetase varia entre 67-100%2,4,5 e está associada a um aumento da morbilidade e mortalidade2-7, sendo o tipo mais frequente a NSIP.6 A patologia intersticial pulmonar pode acometer um início rápido associado a insuficiência respiratória aguda, sendo por vezes refratária ao tratamento.3 No entanto, a maioria dos doentes apresentam um curso crónico e lentamente progressivo.7 As provas de função respiratória demonstram habitualmente um padrão restritivo3-6 e uma alteração da difusão (diminuição da DLCO)5,6; o lavado broncoalveolar demonstra uma alveolite linfocitária, habitualmente CD8+, ou uma alveolite com predomínio de neutrófilos por vezes associada a eosinofilia5; e a tomografia computorizada pulmonar podem revelar um padrão intersticial com vidro de espolido, opacidades lineares, consolidações parenquimatosas, micronódulos com predomínio da região basal e subpleural.3,5,6 Em geral, a miosite precede ou surge concomitantemente com o atingimento pulmonar.3,5 A associação anti-jo1 e anti-Ro é um marcador de envolvimento pulmonar severo.8
Assim sendo, perante uma miopatia inflamatória deverá ser pesquisada a presença do anti-Jo1 e de patologia pulmonar intersticial.3
O fenómeno de Raynaud é precoce em 2/3 dos doentes, podendo preceder a miosite em vários anos.3
Além das manifestações referidas, podem apresentar manifestações gastrointestinais como a disfagia; e cardíacas, sendo a pericardite a manifestação mais frequente.5
A primeira linha de tratamento na síndrome anti-sintetase e na patologia pulmonar intersticial associada a miosite é a corticoterapia em doses altas.1,2,5,7 A dose inicial de prednisolona oral é de 1mg/kg/dia1,2,4,5,7, mas em doentes mais graves pode ser precedida por metilprednisolona 1g/dia durante 3 dias.1,4,7 A taxa de remissão com corticoterapia é de 21% a 68%.4 Apesar disso, a maioria dos doentes necessita de imunossupressão adicional com agentes como a ciclofosfamida, azatioprina, micofenolato de mofetil, ciclosporina, tacrolimus, rituximab ou imunoglobulinas intravenosas.1-5 Normalmente é iniciado um segundo imunossupressor, em sinergia com a corticoterapia, quando existe insuficiência respiratória, patologia severa ou progressiva, ausência de resposta à corticoterapia após 4 a 6 semanas.7 Em casos refratários alguns estudos referem a utilização do rituximab, de pulsos de corticoides e de ciclofosfamida endovenosa. A utilização de terapêutica imunossupressora é baseada apenas na evidência clínica.8
Após um mês de tratamento com corticoterapia, se ocorrer melhoria de sintomas, imagiológica e das provas de função respiratória, deverá ser efetuado um desmame lento para uma duração total de tratamento de 6 a 12 meses, dependendo da resposta à terapêutica.7
Como último recurso, na ausência de resposta à terapêutica, deve ser ponderado o transplante pulmonar.8
A reabilitação pulmonar e a prevenção primária de infeções (vacina da gripe e antipneumocócica) são fundamentais.5
Quanto ao prognóstico, a dispneia em repouso ou para pequenos esforços no momento do diagnóstico é um fator de risco independente associado à mortalidade.5 Outros fatores como a idade avançada, os valores baixos da capacidade vital ou capacidade de difusão; e padrões imagiológicos em favo mel e reticulares implicam um pior prognóstico. Os doentes com vidro despolido apresentam melhor prognóstico.7
A taxa de mortalidade é variável entre 12% a 40%.2,4
Este caso demonstra a importância do diagnóstico e abordagem terapêutica precoces da síndrome anti-sintetase, uma síndrome rara e associada a morbilidade e mortalidade elevadas.
Figura I
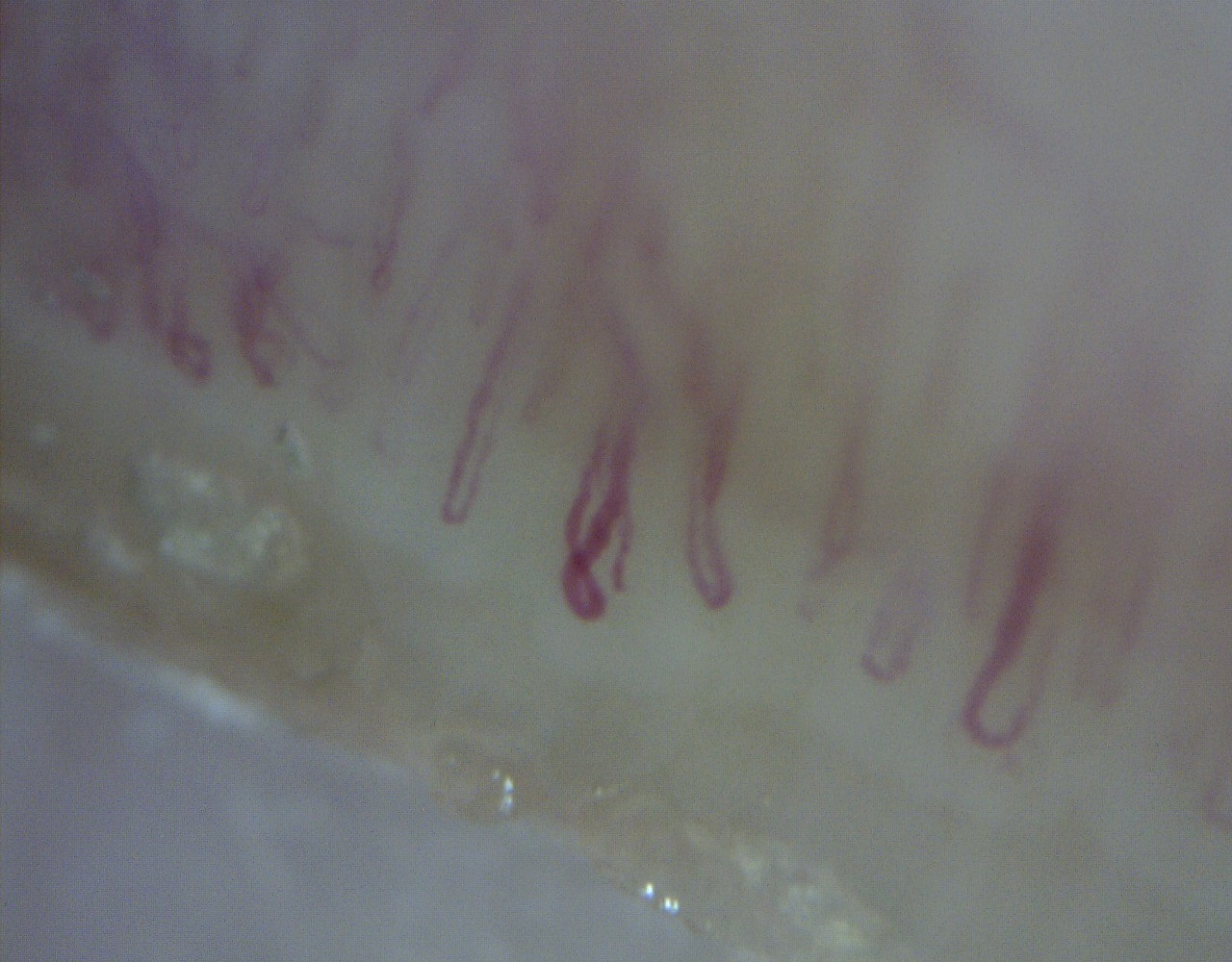
Capilaroscopia onde são observados megacapilares tortuosos e desorganizados e microhemorragias, sem áreas de rarefação.
Figura II
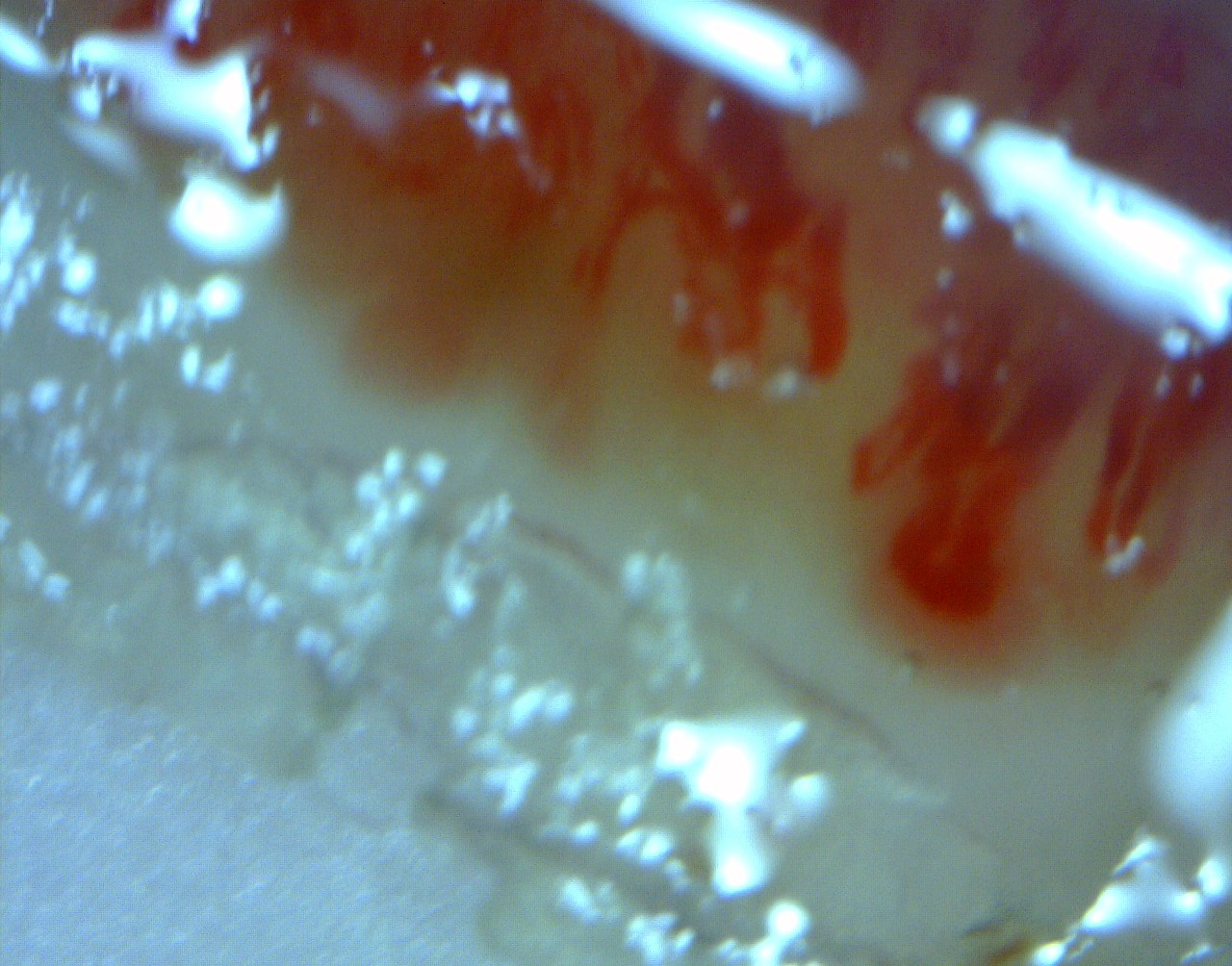
Capilaroscopia onde são observados megacapilares tortuosos e desorganizados e microhemorragias, sem áreas de rarefação.
Figura III
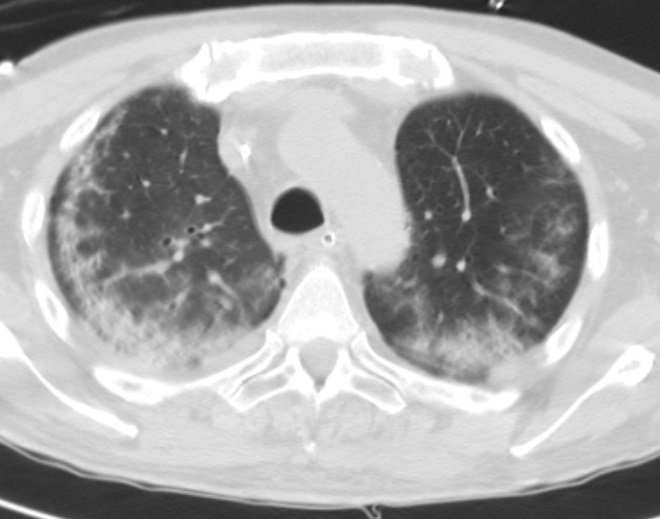
Tomografia computorizada do tórax de alta resolução prévias ao tratamento onde são visualizadas áreas de consolidação parenquimatosa irregular e mal definida na região subpleural posterior dos lobos superiores e inferiores bilateralmente, associadas a vidro de espolido, espessamento peri-bronco-vascular em algumas áreas com preservação de banda parenquimatosa subpleural
Figura IV
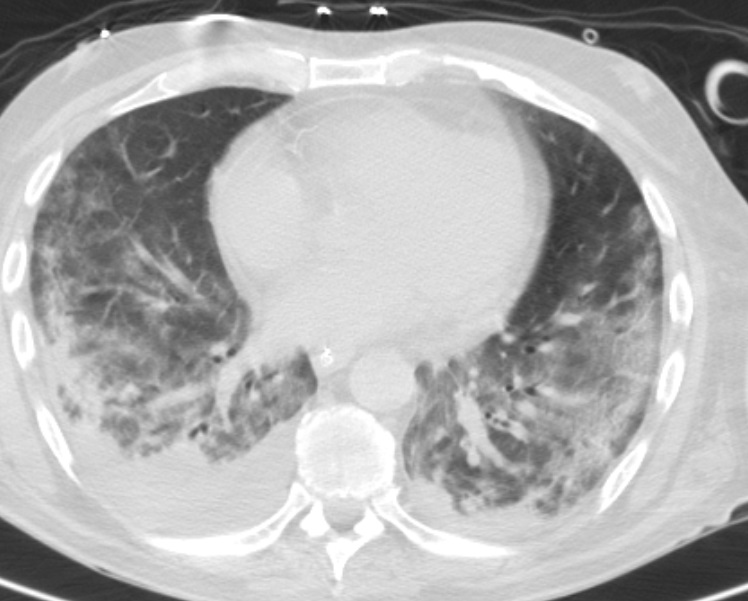
Tomografia computorizada do tórax de alta resolução prévias ao tratamento onde são observadas áreas de consolidação parenquimatosa irregular e mal definida na região subpleural posterior dos lobos superiores e inferiores bilateralmente, associadas a vidro de espolido, espessamento peri-bronco-vascular em algumas áreas com preservação de banda parenquimatosa subpleural
Figura V
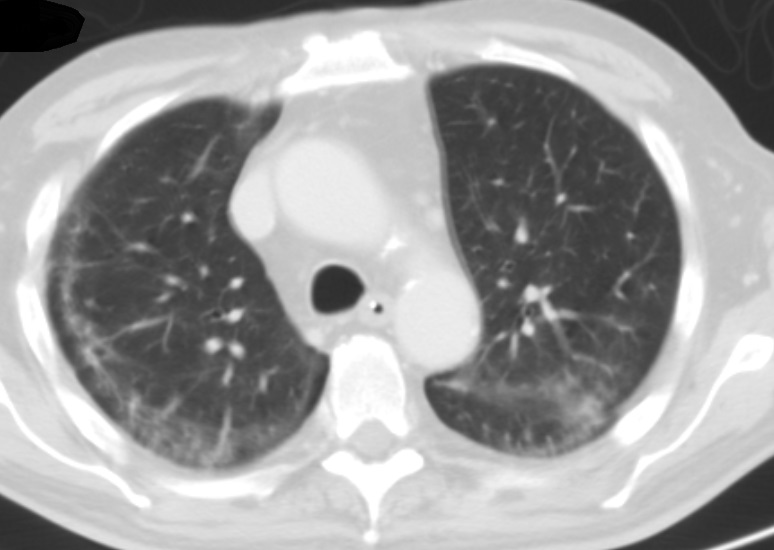
Tomografia computorizada do tórax de alta resolução após 2 semanas de tratamento com corticoterapia onde é observada uma melhoria franca das consolidações pulmonares
Figura VI
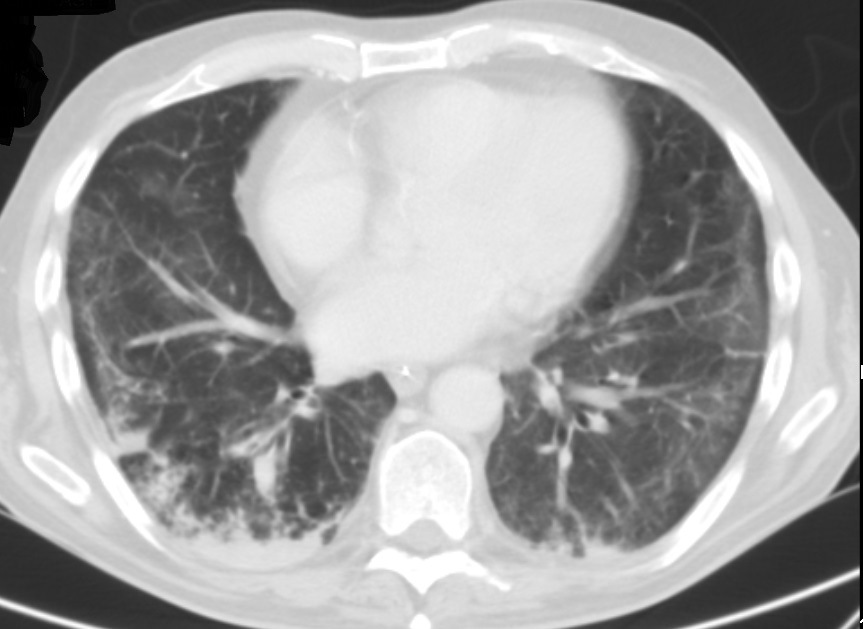
Tomografia computorizada do tórax de alta resolução após 2 semanas de tratamento com corticoterapia onde é observada uma melhoria franca das consolidações pulmonares
BIBLIOGRAFIA
1 - Mammen AL PhD. Autoimmune Myopathies. Continuum. 2016; 22(6): 1852-1870.
2 - Hallowell RW, Danoff, SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies and the antisynthetase syndrome: recent advances. Current Opinion in Reumatology. 2014; 26:684-689.
3 Theilacker LR, Brandão FS, Goulart FV, Vaz JLP, DAlmeida LOD, Salgado MCF. Síndrome Antissintetase: relato de dois casos e revisão da literatura. Rev. Brasileira de Reumatologia. 2015; 55(2):177-180.
4 Shinjo SK, Levy-Neto M. Síndrome antissintetase anti-Jo-1. Rev. Brasileira de Reumatologia. 2010; 50(5): 492-500
5 - Jouneau S, Hervier B, Jutant EM, Decaux O, Kambouchner M, Humbert M, et al. Les manifestations pulmonaires du syndrome des antisynthétases. Review des Maladies Respiratoires. 2015; 32: 618-628.
6 - Dellaripa, PF, Miller ML. Interstitial lung disease in dermatomyositis and polymyositis: Clinical manifestations and diagnosis. 2015 [actualizado em 2015; citado em 2016]. Disponível em www.uptodate.com
7 - Dellaripa PF, Miller ML. Interstitial lung disease in dermatomyositis and polymyositis: Treatment. 2015 [actualizado em 2015; citado em 2016]. Disponível em www.uptodate.com
8 Labirua A, Lundberg IE. Interstitial lung disease and idiopathic inflammatory myopathies: progress and pitfalls. Current Opinion in Reumatology. 2010; 22:633-638.
9 - Targoff IN, MD. Update on myositis-specific and myositis-associated autoantibodies. Current Opinion in Reumatology. 2000; 12:475-481.