Case Report
A 56-year-old male, smoker and without other relevant health history, was admitted to hospital with left anterior chest pain, preceded on the previous week, by fever, myalgia, and fatigue. On admission, physical examination was unremarkable and laboratory testing revealed elevation of acute-phase reactants - c-reactive protein (CPR) 23 mg/dL and erythrocyte sedimentation rate 80 mm/s. On the same day, chest x-ray and CT scan showed interstitial lung fibrosis, mediastinal lymphadenopathy and pericardial effusion, the latter confirmed by transthoracic echocardiography (22 mm, without cardiac tamponade) (Figure 1). He was started on empirical treatment with ceftriaxone and azithromycin. On the next two days, hemodynamic instability and atrial fibrillation developed; pericardiocentesis (380 cc) and electric cardioversion were performed. Pericardial fluid analysis showed an exudate with polimorphonuclear (30%) and mononuclear (70%) cells, microbial studies were negative (Adenosine deaminase, Acid-fast bacilli, culture and viruses). Hemodynamic instability persisted and respiratory failure established, requiring intensive care unit transferral, vasopressor support (noradrenaline) and mechanical ventilation. Transthoracic echocardiography was repeated and showed right ventricular systolic dysfunction (S 9 cm/s), moderated mitral ant tricuspid insufficiency with pulmonary hypertension (PASP 47 mmHg); hemodynamic measurement with Swan-Ganz catheter was compatible with distributive shock, from a presumed respiratory infection. Antibiotics were escalated to piperacillin-tazobactam and vancomycin with good response, accomplishing 14 days of antimicrobial treatment.
At discharge, all tests performed - blood cultures, serology (viruses - B and C hepatitis, Human Immunodeficiency Virus, Cytomegalovirus, Herpes simplex 1/2, Adenovirus, Enterovirus, Parvovirus B19; bacterial - Mycoplasma pneumoniae, Coxiella burnetii, Rickettsia sp, Borrelia burgdorferi, Brucella sp) and autoimmunity (ANA, ENA, anti-cardiolipine, lupus anticoagulant, anti-β2 glycoprotein, anti-dsDNA) were negative. Bronchoalveolar lavage showed normal CD4/CD8 cell count and gallium scintigraphy was negative for active Sarcoidosis.
On clinical re-evaluation, one month later, he was asymptomatic without pericardial effusion and lung fibrosis on CT scan. Six weeks later, he appealed again to hospital with fever and chest pain, and it was found a pericardial effusion relapse with pleural effusion (Figure 2).
At this point, with recurrent febrile serositis attacks and all tests performed (infectious and autoimmunity) negative, the diagnostic hypothesis of Familial Mediterranean Fever (FMF) was considered, and later confirmed by genetic test (heterozygote mutations Arg408Gln and Pro369Ser). He started Colchicine (1 mg/daily) with good tolerance, no recurrence, and no evidence of proteinuria at three years follow-up.
Discussion
First described on 1945 as benign paroxysmal peritonitis (1,2), among populations living around Mediterranean basin (Sephardi Jews, Arabs, Armenians, Turks), later it was also found in other ethnicities (3,4). FMF is a genetic autoinflammatory disease caused by a mutation on MEFV gene, located on 16p13 chromosome, encoding a protein composed of 781 amino acids named pyrin or marenostrin. This protein is almost exclusively expressed on polymorphonuclear leukocytes (PMN), and the best evidence available supports a critical role on regulation of inflammatory process and apoptosis. The proposed pathogenesis for FMF comprises a disruption of marenostrin / pyrin production, due to presence of mutated MEFV gene, allowing microtubular activation and PMN migration, in response of proinflammatory trigger - emotional stress, intense physical activity, temperature extremes and viral infections are suggested, but no clear precipitant is known.
To date more than 300 mutations have been described in Infevers (5), being p. Met694V related to most severe form of the disease and conferring a high-risk of developing amyloidosis. However, there is no straight genotype-phenotype correlation, with great diversity in clinical manifestations of patients carriers of the same mutation, suggesting an important role on environmental factors .
On our case, Arg408Gln and Pro369Ser mutations, both occurring on exon 3, are associated with Armenia and France / Jewish Ashkenazy ancestry, respectively; to our knowledge, no familiar origin association is known.
The first symptoms of FMF usually appear before the age of 20, and only 5% develop the disease after 30. Typical manifestations include recurrent febrile episodes accompanied with serositis, lasting 1-3 days with spontaneous resolution, with leucocytosis and APR elevation; the periodicity is irregular, varying from once a week to once a year. Between crises, FMF patients are asymptomatic. Abdominal pain, with signs of peritonitis, is the most common feature of FMF (8), occurring in around 95% of patients. Joint involvement particularly monoartrhitis of large joints (ankle, knee or hip) is the second most common manifestation. Acute onset pleuritis accompanied with dyspnoea and pleural effusion may occur in up to 50 % of patients (8). Pericardial effusion is rarely described on FMF presentation (3.6% of patients) (9,10), but may be underdiagnosed. The most important long-term complication of FMF, currently less frequent (merits to early treatment) is secondary amyloidosis (AA type).
Despite better recognition of MEFV gene variants, FMF remains a complex clinical diagnosis and various criteria have been proposed with a wide range differences in sensitivity and specificity (11,12). One of the most accepted diagnostic criteria (Figure 3), defines typical attacks by the presence of all of the following features: pain due to serositis, recurrence of attacks (≥ three attacks), fever (≥ 38ºC), and short duration (12h three days). Incomplete attacks are defined by painful and recurrent attacks, not fulfilling all the criteria for a typical attack. According to these criteria, the diagnosis of FMF is established with the presence of: ≥1 major criteria, ≥2 minor criteria, 1 minor plus 5 supportive criteria, or 1 minor plus ≥ 4 of the first five supportive criteria. On our case, we can diagnose FMF with two minor criteria - incomplete chest pain (two attacks) and favourable response to colchicine or one minor criteria (incomplete chest attacks) and four supportive criteria - appropriate ethnic origin (from a Mediterranean country), severe attack requiring bed rest, presence of symptom-free interval, and transient elevation of acute phase reactants.
In relation to the pattern of lung fibrosis present on the first CT scan (and not seen on the second scan), we believe that it was the result of the systemic inflammatory response, due to FMF (with lung involvement) and a superimposed bacterial infection.
Over the last 40 years, colchicine has been the drug of choice for FMF patients. This tricyclic alkaloid, reaches higher concentrations on PMN and acts as microtubule destabilizing agent, thereby, preventing inflammatory response (chemotaxis). Continuous prophylactic therapy prevents both the inflammatory attacks (in 60-65% of patients) and progression towards secondary amyloidosis (2).
Our case represents an atypical presentation of FMF, gathering some particularities: the age at diagnosis, no abdominal or joint involvement, the severity of first attack with superimposed septic shock and the presentation with pleural-pericardial effusion. Thus, a strong clinical suspicion should exist to make a diagnosis of FMF, as this case is illustrative.
Figura I
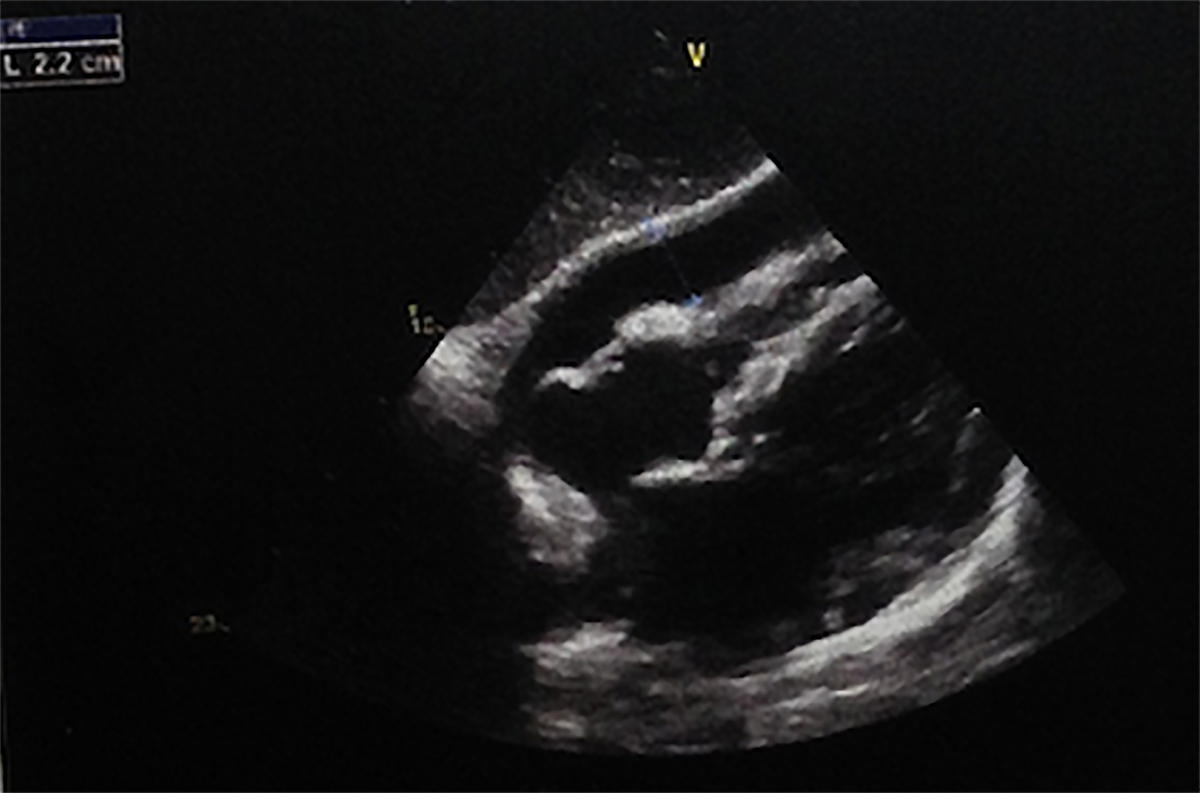
Transthoracic echocardiography showing pericardial effusion (22mm diameter)
Figura II
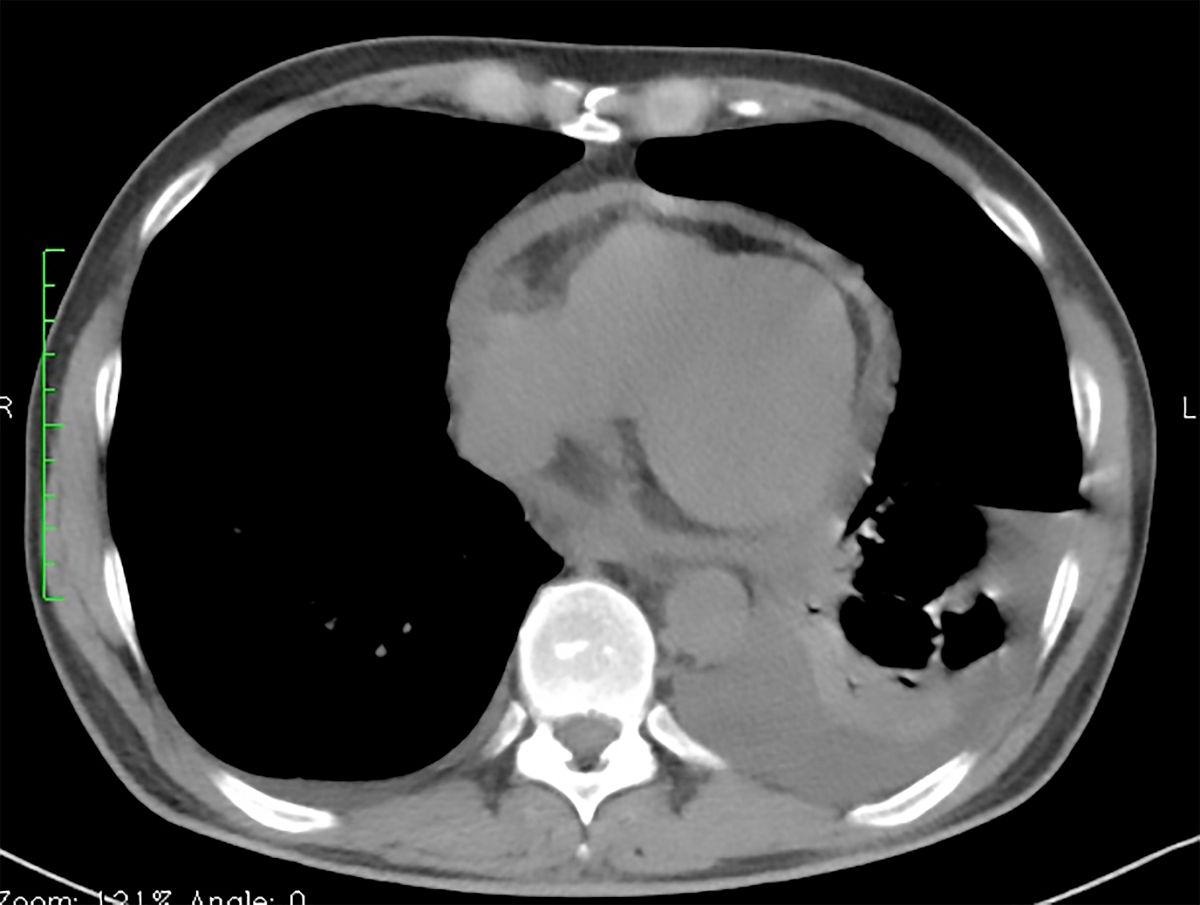
Thorax CT scan (two months and a half after discharge) showing bilateral pleural and pericardial effusion
Figura III
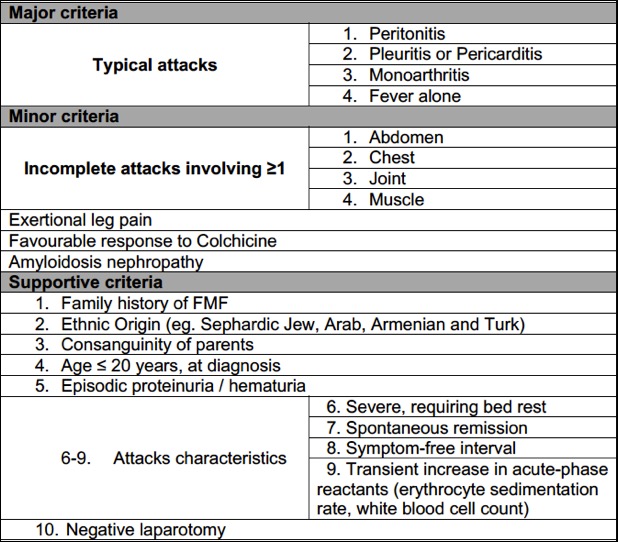
Diagnostic criteria of Family Mediterranean Fever. Adapted from (11)
BIBLIOGRAFIA
1. Gomes JAM, Gomes SM, Conde M. Síndromes Auto-Inflamatórias Hereditárias. Acta Reum Port. 2010;35:14654.
2. Terreri MTRA, Bernardo WM, Len CA, da Silva CAA, de Magalhães CMR, Sacchetti SB, et al. Guidelines for the management and treatment of periodic fever syndromes familial Mediterranean fever. Rev Bras Reum Engl Ed. 2016;56(1):3743.
3. Ben-Chetrit E, Levy M. Familial Mediterranean Fever. Lancet. 1998;351:65964.
4. Padeh S, Berkun Y. Familial Mediterranean fever. Curr Opin Rheumatol. 2016;28.
5. Sarrauste de Menthière C, Terrière S, Pugnère D, Ruiz M, Demaille J, Touitou I. INFEVERS: the Registry for FMF and hereditary inflammatory disorders mutations. Nucleic acids res. 2003 Jan 1;31(1):2825.
6. Kutlay S, Sengul S, Keven K, Erturk S, Erbay B. Two sisters with Familial Mediterranean fever: lack of correlation between genotype and phenotype? J Nephrol. 2006;19(1):1047.
7. Ben-Zvi I, Brandt B, Berkun Y, Lidar M, Livneh A. The relative contribution of environmental and genetic factors to phenotypic variation in familial Mediterranean fever (FMF). Gene. 2012;491(2):2603.
8. Shohat M, Halpern GJ. Familial Mediterranean feverA review. Genet Med. 2011 Jun 23;13(6):48798.
9. Kees S, Langevitz P, Zemer D, Padeh S, Pras M, Livneh A. Attacks of pericarditis as a manifestation of familial Mediterranean fever (FMF). Q J Med. 1997;90(10):6437.
10. Tutar E, Yalçinkaya F, Özkaya N, Ekim M, Atalay S. Incidence of pericardial effusion during attacks of familial Mediterranean fever. Heart. 2003 Sep 15;89(10):1257 LP-1258.
11. Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40(10):187985.
12. Federici S, Sormani MP, Ozen S, Lachmann HJ, Amaryan G, Woo P, et al. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis. 2015 Apr 2;74(5):799 LP-805.

