Introdução
A purpura trombocitopénica trombótica (PTT) caracteriza-se por trombocitopénia, devido à incorporação das plaquetas em trombos na microvasculatura e anemia hemolítica microangiopática, que resulta da destruição dos eritrócitos à passagem pela microcirculação.1
Patologia rara, 1-10 casos por milhão de pessoas por ano.2
Nos doentes com PTT verifica-se a ausência ou deficiência da ADAMTS13 (A Desintegrin And Metallopeptidase with TrombospoSpondin type 1motifs 13th member of the family), que é uma metaloprotease que cliva o fator de von Willebrand. Com essa alteração não há a clivagem dos multímeros do fator de von Willebrand, (tendo por isso maior capacidade adesiva com as plaquetas), levando a sua acumulação à formação de trombos disseminados e a trombocitopenia.1-3
No que respeita a etiologia a PTT pode ser congénita e caracteriza-se por atividade de ADAMTS13 <5% na ausência de anticorpos anti ADAMTS13; ou adquirida que é a forma mais comum e trata-se de uma doença autoimune com anticorpos anti ADAMTS13. A PTT adquirida é na maior parte das vezes classificada com idiopática dada a dificuldade do diagnóstico etiológico, no entanto são conhecidas algumas associações com a infeção por HIV, gravidez, doenças neoplásicas, drogas. 4-6
A apresentação clinica é descrita classicamente pela pentade: febre, disfunção renal, trombocitopenia, alterações neurológicas e anemia hemolítica microangiopática. No entanto a sua especificidade e sensibilidade são baixas. As alterações laboratoriais precoces como a trombocitopenia e os marcadores de hemólise permitem o diagnóstico atempado de uma condição potencialmente fatal na ausência de tratamento precoce e adequado.2,5,6
A plasmaférese é uma técnica de filtração de plasma que alterou verdadeiramente o prognóstico da PTT, com a descoberta da técnica a mortalidade passou de cerca de 90% para 10-20%1,3. Embora, sempre que disponível, seja a primeira linha de tratamento da PTT adquirida, existe nas recomendações da British of Hematology Journal a exceção da PTT relacionada com doenças malignas, onde, tendo por base os mecanismos fisiopatológicos conhecidos, está apenas recomendado o tratamento da doença de base4.
Caso Clinico
Sexo masculino, 53 anos de idade, sem antecedentes pessoais ou familiares de relevo e sem terapêutica habitual. Recorre ao serviço de urgência referindo episódio de parestesias da mão e hemiface esquerdas e disartria transitória. Negava sintomas respiratórios, urinários ou gastrointestinais. Sem clinica sugestiva de infeção. Negava consumo recente de fármacos. Sem intercorrências infeciosas recentes.
À admissão no serviço de urgência o doente estava hemodinamicamente estável, exame neurológico sem alterações e do restante exame físico de destacar palidez da pele e mucosas, sem sinais de discrasia hemorrágica, e adenopatia axilar direita com cerca de 2cm, móvel, de consistência elástica, contornos bem definidos e indolor a palpação. Do estudo analítico realizado à admissão (Tabela 1): trombocitopenia (17000/uL); sem alterações da coagulação; anemia (hemoglobina 6,3g/dl) com critérios de hemólise. Teste de Coombs negativo; lesão renal aguda com creatinina 1,71 mg/dL. Realizou também tomografia computorizada (TC) de craneo que não mostrava alterações agudas e TC toracoabdominopelvica que revelou adenopatias generalizadas (axilares, hilares, retroperitoneais de tamanho < 1cm e uma pélvica com 4cm) e esplenomegalia ligeira (145mm) heterogénea sugerindo possíveis microenfartes (Fig. 1a e Fig. 1b).
Com os dados clínicos e dos exames complementares de diagnóstico foi possível estabelecer o diagnóstico de PTT de etiologia não esclarecida, mas provavelmente secundária.
Foi complementada a investigação etiológica: Biópsia óssea: hipercelularidade marcada com fibrose colagénea sem células ectópicas, sugestiva de mielofibrose; Mielograma: hiperplasia eritróide nas particulas ósseas; Imunofenotipagem medular: sem alterações; Estudo autoimune: negativo; e Eletroforese sérica de proteínas: sem alterações de relevo; serologias viricas negativas; biópsia da adenopatia axilar protelada dado o risco hemorrágico.
A par da investigação ao 3º dia de internamento iniciou plasmaférese com reposição de 4,4L de plasma fresco congelado (PFC, Cálculo = (0,065xpesoKg(93Kg)) x (1-hematócrito)). Na 3ª sessão apresentava recuperação completa da função renal e 59000 plaquetas, tendo por isso iniciado ácido acetilsalicílico (AAS) 100mg/dia. Passou a sessões em dias alternados e na 6ª sessão atingiu as 108000 plaquetas. Decidido aumentar o intervalo das sessões para dois dias e na 8ª sessão apresentava 124000 plaquetas, altura em que suspendeu a técnica. A atitude de suspender a técnica teve por base o desconhecimento do verdadeiro mecanismo desta PTT e a presunção de se tratar de uma PTT secundária a patologia maligna.
Ao quarto dia após ter suspendido a plasmaferese (precocemente, antes de atingir as 150000 plaquetas) era evidente novo agravamento da trombocitopenia (57000/microlitro) e da anemia hemolítica (Hb 11,48g/dl, LDH 302 U/L, Haptoglobinas 18mg/dl e esquizócitos do ESP). Retomou plasmaferese e na 11ª sessão (3ª deste segundo ciclo) iniciou prednisolona 1mg/Kg/dia. Após melhoria inicial da trombocitopenia na 20ª sessão, novo agravamento da trombocitopenia mas microangiopatia controlada sugerindo que naquela altura não seria apenas a PTT a condicionar a trombocitopenia. Alargado o estudo etiológico até a data inconclusivo: pesquisa JacK 2 negativo; marcadores tumorais normais; doseamento de ADAMTS13 e Ac ADAMTS13 que revelou presença de Ac com título e diminuição da actividade de ADAMTS13, compatível com PTT adquirida; ao exame objectivo não havia qualquer adenopatia palpável pelo que repetiu TC que confirmou a diminuição da adenopatia axilar, impossibilitando a biópsia e a esplenomegalia com baço heterogéneo. Sem diagnóstico etiológico possível após toda a investigação e doença não controlada com terapêutica otimizada e plasmaferese diária, apesar do risco hemorrágico (35000 plaquetas) optou-se por esplenectomia. Avaliado o risco hemorrágico vs trombótico de transfundir plaquetas num doente com PTT, optou-se pela transfusão de um pool de plaquetas. O procedimento decorreu sem intercorrências e o exame histológico mostrou envolvimento esplénico e de 3 gânglios linfáticos do hilo esplénico por processo linfoproliferativo com características de Linfoma Hodgkin clássico de tipo celularidade mista.
Optou-se por um esquema que incluisse corticoide e que permitisse a interrupção com menor risco de pancitopenias pelo que acabou por iniciar LVPP (clorambucil, vinblastina, procarbazina e prednisolona), que teve de suspender ao 9º dia por neutropenia. Fez G-CSF e infusões de PFC até reunir condições para novo tratamento com outro esquema: ABVD (doxorrubicina, bleomicina, vinblastina e dacarbazina) que cumpriu durante 8 sessões com remissão completa da doença. Aos 24 meses de follow-up o doente encontra-se assintomático sem qualquer episódio de PTT ou sinal de doença de Hodgkin ativa.
Discussão:
A Anemia Hemolítica microangiopática relacionada com patologia maligna é uma entidade rara com uma incidência de cerca de 0,25-0,45 pessoas/milhão e ainda mais rara é a sua expressão com critérios de PTT. Habitualmente está associada a tumores sólidos, mestastizados e comporta-se mais vezes como coagulação intravascular disseminada (CID) do que como verdadeira PTT7. A fisiopatologia é pouco clara. Um dos mecanismos parece ser a fragmentação/destruição dos glóbulos vermelhos e plaquetas na passagem pelos vasos dos tecidos neoplásicos. Há ainda referência ao papel das citoquinas produzidas pelo tumor com actividades diversas ainda pouco compreendidas5,7-9.Está descrita também a diminuição não significativa da actividade da ADAMTS13 mas não a presença de Ac anti ADAMTS13.
A PTT secundária a doença de Hodgkin (e provavelmente secundária a outros linfomas) é então fisiopatologicamente uma entidade muito mais relacionada com a PTT associada ao HIV ou drogas do que com a PTT associada a doença maligna. Neste doente, a presença de Ac justifica a resposta à plasmaférese (ao contrário do que referem as recomendações do tratamento da PTT associada a doença maligna4) e o agravamento da trombocitoponeia dissociado da microangiopatia.
Sendo a fisiopatologia ainda mal entendida as recomendações sobre o tratamento são escassas. A British Journal of Hematology nas suas últimas recomendações de 2012 refere apenas que a plasmaférese não está indicada e que o tratamento da doença de base tem de ser considerado4. No caso descrito a trombocitopenia grave e progressiva de uma PTT de etiologia não esclarecida obrigou a múltiplas sessões de plasmaférese (gold standarde técnica life saving na terapêutica da PTT3-5) com melhor resposta do que a perfusão de PFC, o que o resultado positivo da pesquisa de Ac anti ADAMTS13 veio justificar. Das terapêuticas coadjuvantes descritas AAS, corticoide, rituximab, ciclosporina5, foram apenas utilizados a AAS e prednisolona sem melhoria clinica observada após a sua introdução. A esplenectomia também com valor cada vez mais questionável na terapêutica da PTT4-6, veio neste caso ter um valor acrescido como meio de diagnóstico. O tratamento da doença de base, mostrou-se exequível e de primordial importância.
As revisões da literatura referem que existem apenas 14 casos descritos de associação com linfomas e apenas 4 casos de linfoma de Hodgkin7. A falta de suporte em experiência prévia representou um obstáculo na condução do diagnóstico e do tratamento. Fazendo uma avaliação critica e criteriosa da marcha diagnóstica, impõe-se refletir sobre a importância do diagnostico etiológico, (pois só o tratamento direcionado permitiu controlar a PTT), e sobre o provável excesso de zelo no adiamento da exérese de um gânglio pelo risco hemorrágico que foi incontornável numa fase posterior face à investigação inconclusiva.
Em conclusão a PTT relacionada com doenças do tecido linfoide não deve ser entendida como PTT secundária a doenças malignas pois os mecanismos, como ilustra este caso, podem ser distintos, necessitando por isso de uma abordagem diferente e no que diz respeito ao diagnóstico e ao tratamento o doseamento da atividade da ADAMTS13 e a plasmaferese têm um papel incontornável.
Agradecimentos:
Os autores agradecem as contribuições inestimáveis da Dra Margarida Inácio, Dra Marisol Guerra e Dra Ana Carvalho e ao Dr. Rui Costa na discussão crítica do caso clínico e na revisão do seu manuscrito.
Quadro I
Resultados analíticos realizados à admissão
| | | |
| | | Valores de Referência |
| Hb | 6,3 g/dl | 13,0 - 18,0g/dl |
| Plaquetas | 17000/microlitro | 150-400/microlitro |
| ESP | Esquizócitos | |
| Reticulócitos | 17,3% | 11-16% |
| Haptoglobinas | 18mg/dl | 25-190mg/dl |
| Teste de Combs | Negativo | |
| PT e aPTT | Normais | |
| D-Dimeros | 0,3 ug/mL | <0,5ug/mL |
| Ureia | 82 mg/dL | <50mg/dL |
| Creatinina | 1,71 mg/dL | 0,7-1,4mg/dL |
| LDH | 970 UI/L | 135-225UI/L |
| BilirrubinaTotal | 1,7 mg/dL | <1,2mg/dL |
| Troponina I | 0,2 mg /L | <0,1mg/dl |
| Função Tiroideia | Normal | |
| Anti-HIV | Negativo | |
| Anti-HCV e HBV | Negativos | |
| Paul Bunnel | Negativo | |
| Índice reticulocitário | 10 | |
Figura I
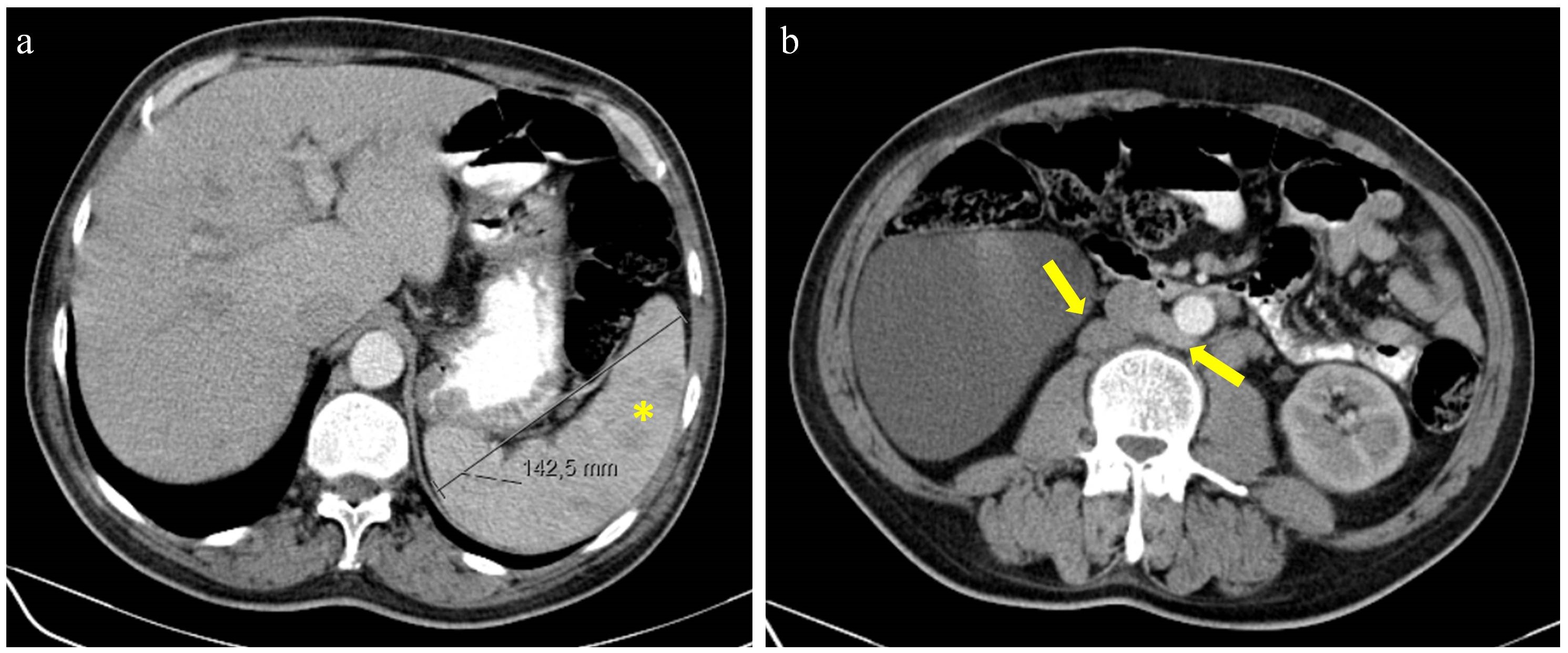
TC abdominal. (a) esplenomegalia (asterisco); (b) múltiplas adenopatias retroperitoneais infracentimétricas (setas).
BIBLIOGRAFIA
1.(1) Fogarty PT et al. Current Medical Diagnosis & Treatment, 2014.
2.(2)Pessegueiro P, Pires C. Sindrome hemolítico urémico/Purupura trombocitopénica trombótica. Revista da Sociedade Portuguesa de Medicina Interna 2005 102 116.
3.(3)Bombery P, Scully M. Management of thrombotic thrombocytopenic purpura: current perspectives. Journal of Blood Medicine 2014:5 15-23
4.(4)Scully M, Hunt BJ, Benjamim S, et al. Guidlines on the diagnosis and management of thrombotic thrombocytopenic purupura and other thrombotic microangiopathies. British Journal oh Hematology 2012.
5.(5)Coppo P, Veyradier A. Current management and therapeutical perspective in thrombotic thrombocytopenic purpura. La Presse Medicale 2012, 41: e163 e176.
6.(6)George J. How I treat patients with thrombotic Thrombocytopenic Purpura. Blood 2010 4060-4069.
7.(7)Lechner K, Obermeier H. Cancer Related Microangiopathic Hemloytic Anemia, Clinical and Laboratory Features in 168 Reported Cases. MD Journal. 2012 ; 91: 00 00
8.(8)Blot E, Decaudin D, Veyradier A, Bardier A, Cancer related thrombotic microangiopathy secondary to Von Willebrand factor cleaving protéase deficiency. Thromb Res. 2002; 106: 127 130
9.(9)Elliott MA, Letendre L, Gastineau DA, Winters JL, Pruthi RK, Heit JÁ: Cancer associated microangiopathic hemolytic anemia with trombocytopenia: a importante diagnostic consideration. Eur J Haematol. 2010; 85: 43 50

