

Chronic myeloproliferative disorders are a subgroup of the haematological neoplasms of the myeloid series. Among these, chronic myeloid leukaemia (CML), polycythemia vera (PV), primary myelofibrosis (PMF) and essential thrombocythemia (ET) are the ones currently best characterized.
ET, unlike CML, PV and PMF, is often an exclusion diagnosis in the presence of chronic thrombocythemia of a non-reactive nature. The presence of JAK2, MPL and CALR gene mutations, found in about 90% of the patients with ET, have come to be of important aid in the diagnostic certainty for these patients.[1]
ET is often asymptomatic and discovered incidentally when thrombocytosis is noted on a complete blood count obtained for some other reason. The main clinical manifestations are vasomotor symptoms and thrombohemorrhagic complications. Digital ischemia is a rare complication, which may start as the Raynaud phenomenon (RP) and progress to ischemic necrosis of the terminal phalanges.
Case Presentation:
A 48-year-old female patient was referenced to the urgent care department, with a necrosis in the third toe of the left foot (Figures 1 and 2) and second and third toes of the right foot (figures 3 and 4). She is an active smoker, with no other relevant personal medical history. About 10 months earlier she had been admitted to another hospital because of inaugural pain and darkening with progressive worsening of 3 toes in the right foot. After etiologic investigation and exclusion of vascular aetiology of atheroembolic nature, RP was admitted and nifedipine was started. When observed in a vascular surgery consultation, the thrombophilic test revealed rheumatoid factor (RF) 99 UI/mL, antinuclear antibodies (ANA) positive (1/160), and positive lupus anticoagulant. It was decided to initiate a low molecular weight heparin, with subsequent symptomatic improvement and scarring of the digital lesions. Four months later, a new relapse of the lesions occurs, with an extension to the third toe of the left foot, with severe and persistent pain. She was hemodynamically stable and peripheral pulses were palpable, ample and symmetric. The patient presented an area of distal necrosis in the second and third toes of the right foot, first and the second toe of the left foot, with the presence of livedo reticularis (Figures 1-4). Further complementary exams revealed leukocytosis (16,7x109/L) with neutrophilia (80%), thrombocytosis (650.000x109/L), with a normal hemoglobin level (13.9 g/dL) and no identifiable alterations on blood smear. The chest radiograph and electrocardiogram were normal. The patient was hospitalized for etiological investigation. The laboratory study was negative for ANA, antineutrophil cytoplasmic antibodies, anti-dsDNA antibodies, anti-smith, anti-ribonucleoprotein, anti-Scl 70, anticentromere, antiphospholipid antibodies, with slightly elevated RF (41 UI/L) and normal complement factor levels. The search for cryoglobulins was negative, protein C and S levels were normal and no activated protein C resistance was shown. Infectious serology was negative for syphilis, human immunodeficiency virus, hepatitis B and C virus. Proteinuria in 24h urine was 224mg. A thoracic-abdominal-pelvic computed tomography (TAP-CT) excluded the existence of adenomegalies or any other relevant changes, and the absence of a macrocirculatory dysfunction was confirmed by a Doppler study of the inferior members. A bone marrow biopsy and aspiration were performed, which revealed: evidently enlarged and multiple megakaryocytes, with irregular forms and the formation of cellular aggregates. The cytogenetic study was normal but the mutation analysis showed the presence of the JAK2-V617F mutation and the absence of the BCR/ABL translocation, compatible with the ET diagnosis. During hospitalization, there was a global clinical improvement with the introduction of systemic vasodilators, antiaggregation, and analgesia, leading to a greater delimitation of the digital necrotic area and pain control. The patient was discharged with aspirin and smoking cessation. In the first follow-up appointment she manifested a partial symptomatic relief, analytically with persistent thrombocytosis (705x109/L), and for these reasons, it was started hydroxyurea 1g/day. After 2 months of cytoreductive therapy, she had a platelet count of 224x109/L, with substantial improvement of the digital necrotic lesions.
Discussion: The authors describe the case of a patient with digital ischemia, whose differential diagnosis implies exclusion of macrocirculatory aetiology (atheroembolic or as peripheral arterial disease), secondary RP and associated connective tissue diseases, thrombophilias, thrombotic microangiopathy, disseminated intravascular coagulation (DIC), myeloproliferative syndrome, obliterating thromboangiitis and ET. ET and obliterating thromboangiitis are both a diagnosis of exclusion, which hampers differential diagnosis between these two entities. Computed angiography of the inferior extremity arterial system excluded a macrocirculatory aetiology. Main causes of secondary RP were ruled with the negative screening for autoantibodies. Viral serologies were also negative. The hypothesis of thrombophilia was excluded because of the normal dosing of natural anticoagulants, negative antiphospholipid antibodies and the absence of resistance to activated protein C. Thrombotic microangiopathy diagnosis was unlikely in the absence of microangiopathic hemolytic anaemia, normal peripheral blood smear and thrombocytosis. Coagulation tests ruled out DIC. In order to investigate the existence of a myeloproliferative disorder, a TAP-CT evaluation was solicited, which excluded the existence of splenomegaly or adenomegalies. The definitive exclusion of this aetiology and the existence of a persistent thrombocytosis pattern led to a bone marrow biopsy and aspiration. The V617 mutation of the JAK2 gene has been detected and BCR/ABL translocation was absent. These results were compatible with ET and ruled out obliterating thromboangiitis hypothesis. ET is described in the literature as a rare cause of digital ischemia, which should be excluded in all patients with otherwise unexplained digital ischemia.[2]
ET is diagnosed in the presence of a persistent thrombocytosis and in the absence of a reactive cause (table 1) or other myeloid neoplasms. With an estimated incidence of 1-2.5 new cases/100.000 patients/year and a prevalence of 24/100.000, this pathology is more common in female individuals and among Afro-Americans. The average age at diagnosis is 60, with about 20% of patients diagnosed under 40. Patients with ET and JAK V617F mutation (50%-64%) generally have a higher propensity for thrombotic events and PV progression.[1, 3-9] However, it is important to recognize that this mutation only allows to set apart patients with ET from those with reactive thrombocytosis, not allowing the differentiation between ET, PV and PMF.[1] A normal haemoglobin value in the absence of an iron deficit usually allows the exclusion of a PV diagnosis. PMF can usually be excluded in the absence of significant splenomegaly, anaemia, dacrocytes and leukoerythroblastosis.[10] Nowadays the diagnosis of ET by the 2016 World Health Organization (WHO) criteria (table 2) requires all four of the major criteria or three major criteria plus the minor criterion.
In about 50% of the cases of ET, there are no clinical manifestations at the time of the diagnosis.[1] Those who have symptoms refer mainly vasomotor symptoms (headache, syncope, atypical precordialgia, acral paresthesia, erythromelalgia, livedo reticularis or visual disturbances) or thrombohemorrhagic complications due to qualitative and quantitative platelet changes. Frequent thrombotic incidents include stroke, retinal vessel occlusion, coronary ischemia, venous thromboembolism, hepatic or portal vein thrombosis and, more rarely, digital ischemia. Digital ischemia may present itself initially as RP that can progress to a necrosis of the digital phalanges, such as noted in this patient.[1] Hemorrhagic events are usually more frequent in patients with platelet counts above 1 million, aspirin or nonsteroidal anti-inflammatory drugs therapy.
The main goal of treatment of ET is to prevent thrombotic and hemorrhagic complication and to alleviate symptoms.[11] The International Prognostic Score of Thrombosis in WHO-essential thrombocythemia (IPSET-Thrombosis) score stratifies the risk of thrombotic complications as follows:
- High-risk: History of thrombosis at any age and/or age> 60 years with a JAK2 V617F mutation;
- Intermediate-risk: Age > 60, no JAK2 mutation detected, and no history of thrombosis;
- Low-risk: Age ≤ 60 with JAK2 mutation and no history of thrombosis;
- Very low-risk: Age ≤ 60, no JAK2 mutation detected, and no history of thrombosis.
Hydroxyurea (HU) is the best-known agent and the first-line treatment for most high-risk patients, and it has proven to reduce the number of thrombotic events on randomized control trials.[12] As an alternative cytoreductive agent, there is anagrelide, considered to be efficient, but there is a higher rate of therapy withholding due to its adverse effects.[13-15]
In addition to cytoreduction, patients should be treated with systemic anticoagulation and/or an antiplatelet as follows:
- History of Venous Thrombosis: Long-term HU plus systemic anticoagulation;
- History of Arterial Thrombosis: Long-term HU plus low dose aspirin (LDA) < 100 mg)
- Age > 60 and no history of venous or arterial thrombosis: recommendation to HU plus aspirin is stronger if JAK2 V617F mutation is present.
Cytoreductive therapy is not recommended for most of the patients with the low and very low-risk disease. One accepted approach is as follows:
- Presence of vasomotor symptoms - Daily LDA
- Both JAK mutation and cardiovascular risk factors - Twice daily LDA
- Either JAK2 V617F mutation or cardiovascular risk factors - Daily LDA
- Neither JAK2 V617F mutation, cardiovascular risk factors or vasomotor symptoms - Observe without treatment
Most patients have a normal life expectancy and should be advised to discontinue smoking, control obesity, and other cardiovascular risk factors.
Quadro I
Reactive Thrombocytosis Causes
| Reactive Thrombocytosis Causes | |
| Nonmalignant hematologic disorder | Acute hemorrhage; Hemolytic anemia; Iron deficiency; Vitamin B12 deficiency treatment; Rebound effect after immune or ethanol-induced thrombocytopenic purpura treatment |
| Malignant disease | Metastatic neoplasm; Lymphoma; Rebound after myelosuppression |
| Acute or Chronic Inflammatory Disease | Autoimmune diseases; Inflammatory bowel disease; Celiac disease; Surgical or functional asplenia |
| Infections | |
| Tissue injuries | Acute Myocardial Infarction; Burns; Severe trauma; Acute Pancreatitis; Post-operative |
| Chronic Kidney Disease | |
| Severe Reactions to Medication | Vincristine; Corticosteroids; Epinephrine; Interleukin 1B; Retinoic acid; Low molecular weight heparin |
Quadro II
ET Diagnostic Criteria (WHO 2016)
| ET Diagnostic Criteria (WHO 2016) | |
| Major Criteria | Thrombocytosis >450.000 mcg/L |
| Major Criteria | Bone marrow biopsy with proliferation of megakaryocyte lineage with increased numbers of enlarged, mature megakaryocytes with hyperlobulated nuclei, no significant increase or left shift in neutrophil granulopoiesis or erythropoiesis and very rarely minor (grade 1) increase in reticulin fibers |
| Major Criteria | WHO criteria for BCR-ALB1 positive chronic myeloid leukemia, polycythemia vera,primary myelofibrosis, myelodisplastic syndrome, or other myeloid neoplasm not met |
| Major Criteria | Demonstration of a JAK2, CALR, or MPL mutation |
| Minor Criteria | Demonstration of another clonal marker (ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, or SR3B1 mutation) or no identifiable cause of thrombocytosis (eg. Infection, inflammation, iron deficiency anaemia) |
Figura I
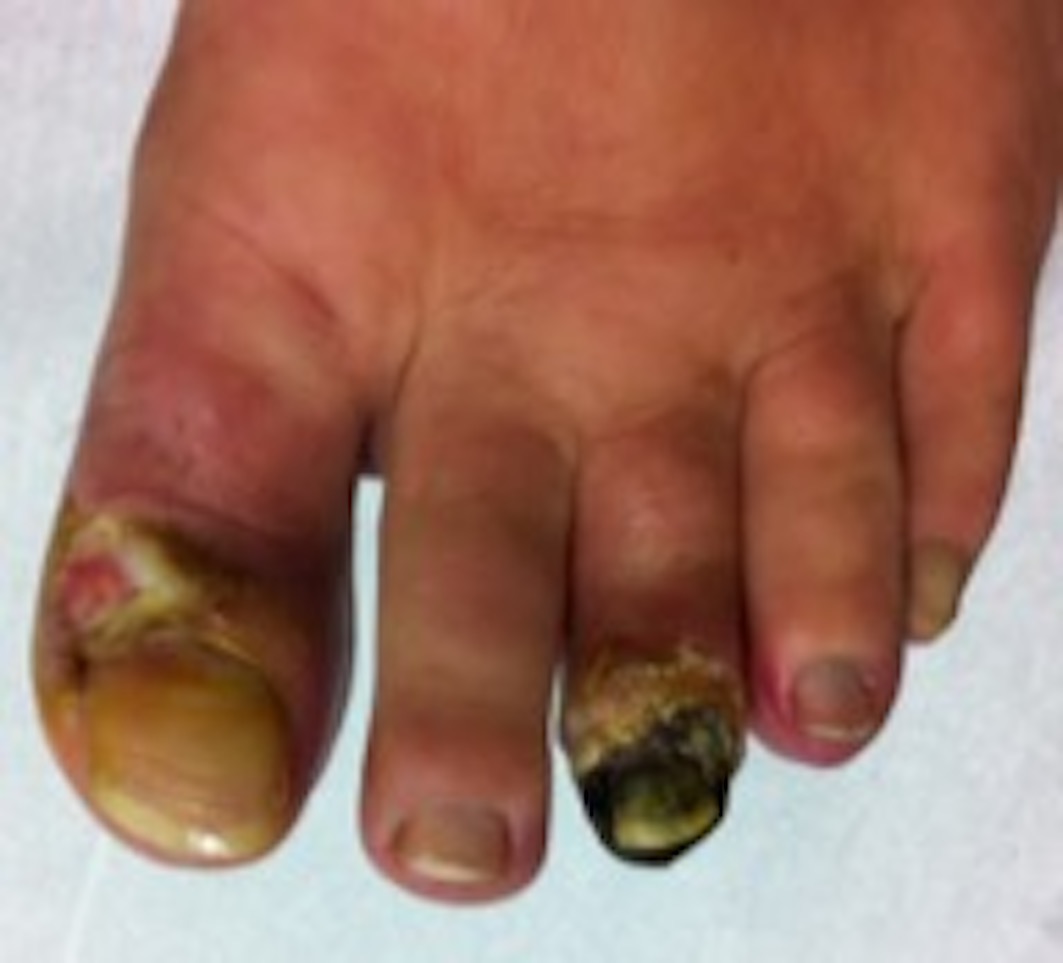
Distal digital necrosis
Figura II
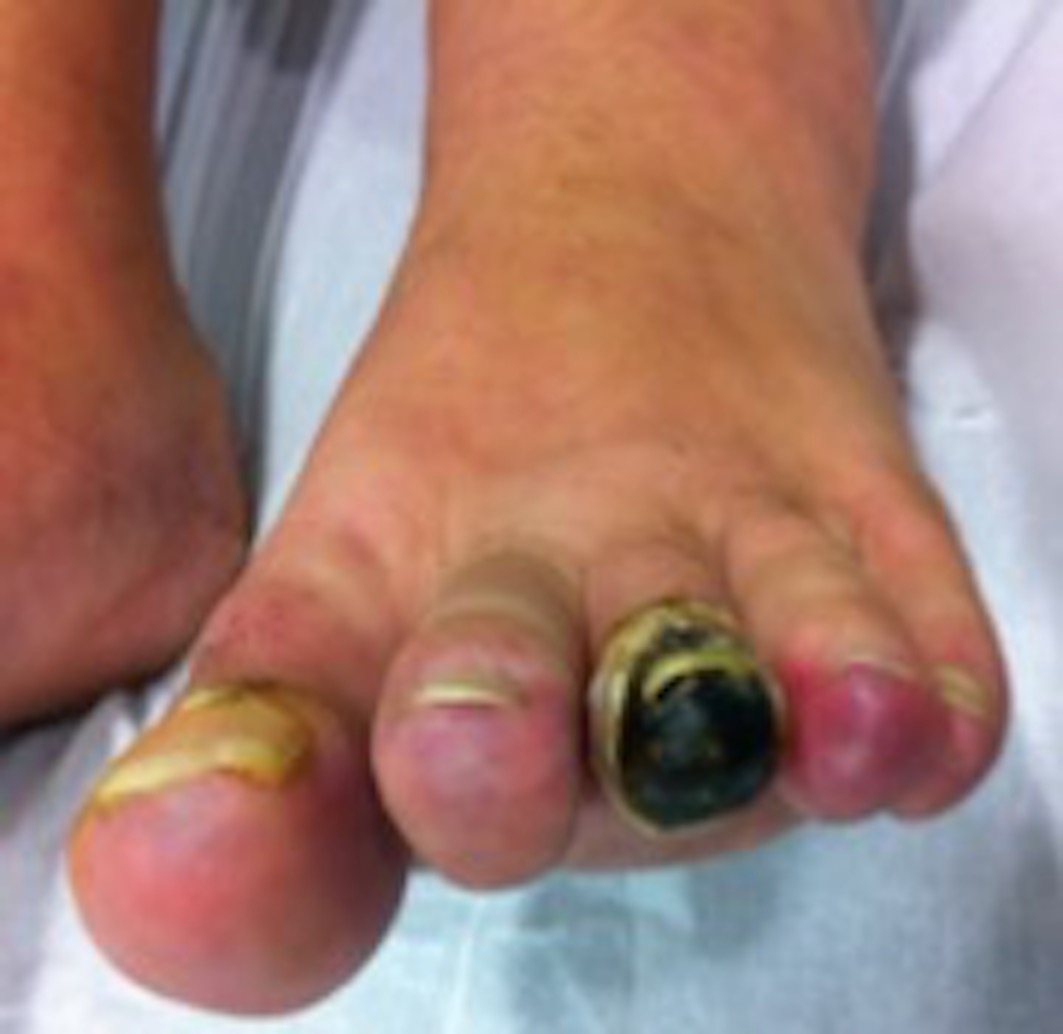
Distal digital necrosis
Figura III
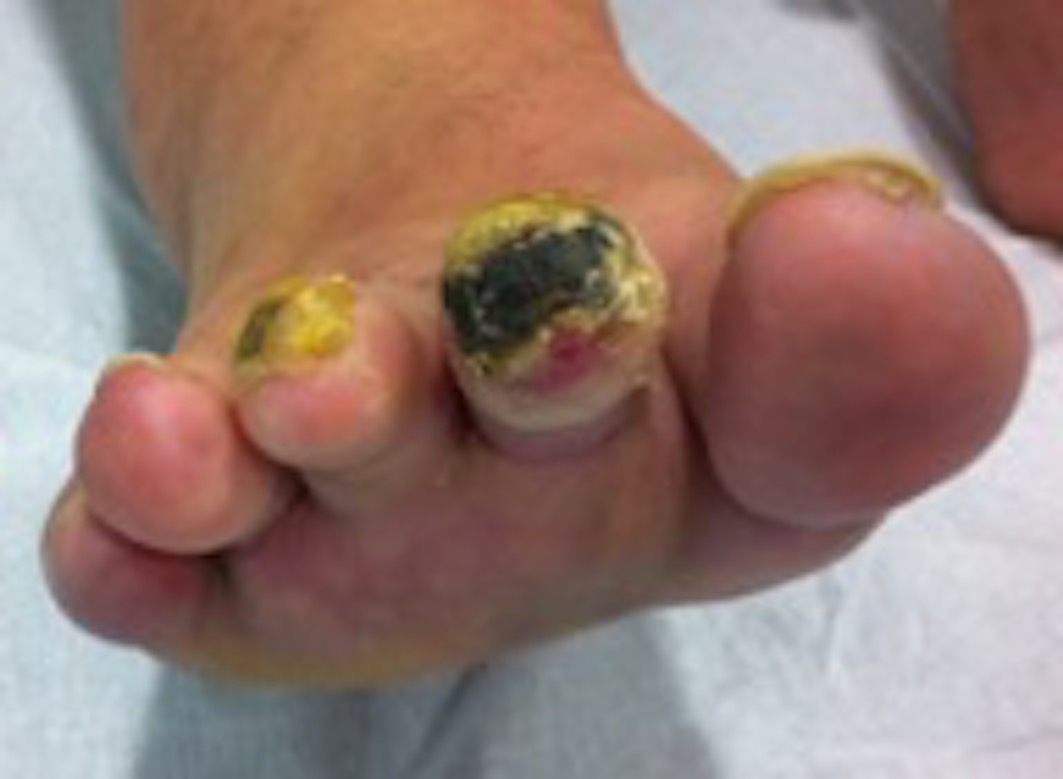
Distal digital necrosis
Figura IV
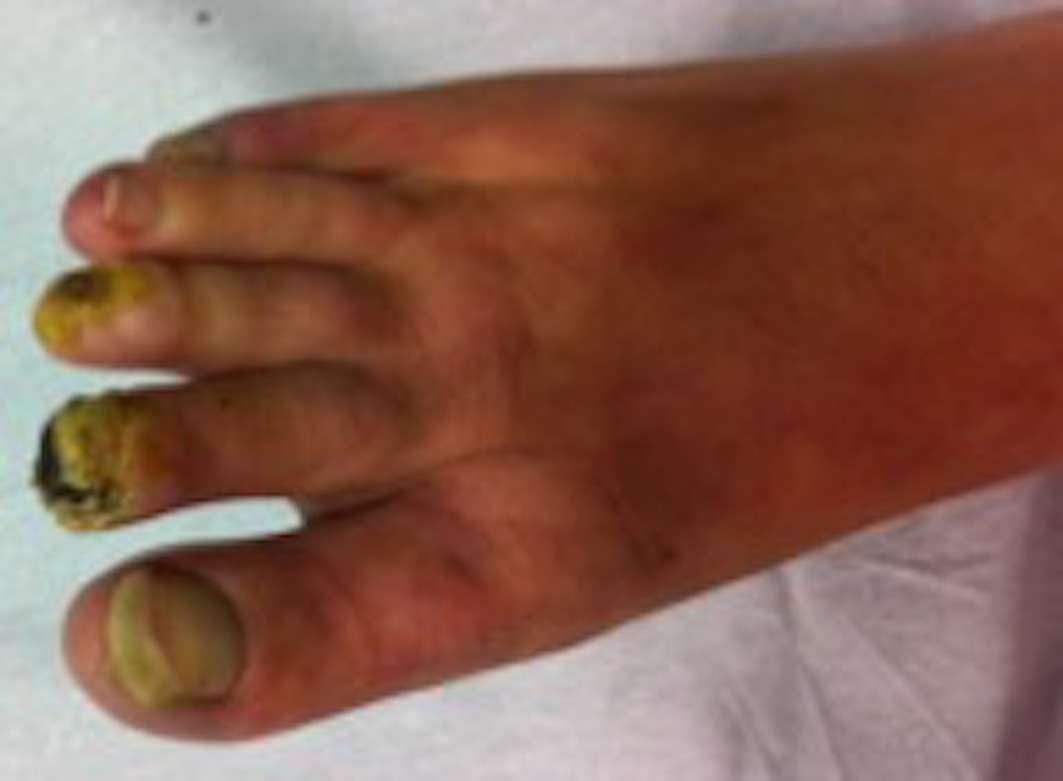
Distal digital necrosis
BIBLIOGRAFIA
1 - Tefferi A. Diagnosis and clinical manifestations of essential thrombocythemia. In UpToDate, Post, TW (Ed). UpToDate, Waltham, MA, 2017
2 - Papadonikolakis A, Chloros GD, Smith, BP, Koman, LA. Digital Ischemia Due to Essential Thrombocythemia: A Case Report. J Hand Surg Am. 2007 Sep; 32(7):1053-7
3 - Schafer A. Molecular basis of the diagnosis and treatment of polycythemia vera and essential thrombocythemia. Blood, 2006; 107(11):4214
4 - Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT, et al. Disorders Study Group, Medical Research Council Adult Leukaemia Working Party, Australasian Leukaemia and Lymphoma Group. Lancet, 2005; 366(9501):1945
5 - Gangat N, Wolanskyj AP, McClure RF, Li CY, Schwager S, Wu W, Tefferi. Risk stratification for survival and leukemic transformation in essential thrombocythemia: a single institutional study of 605 patients. Leukemia. 2007;21(2):270.
6 - Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544-1551
7 - Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. 2013; 123(10):1552-5
8 - Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia clinical associations and long-term prognostic relevance. Br J Haematol 2005, 131:208
9 - Kittur J, Knudson RA, Lasho TL, et al. Clinical correlates of JAK2V617F allele burden in essential thrombocythemia, Cancer 2007; 109:2279
10 - Beer PA, Erber WN, Campbell PJ, Green AR. How I treat essential thrombocythemia. Blood. 2011; 117(5):1472-1482
11- Tefferi A, Rosmarin. Prognosis and treatment of essential thrombocythemia. In UpToDate, Post, TW (Ed). UpToDate, Waltham, MA, 2017
12 - Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995; 332(17):1132-1136
13 - Murphy S, Iland H, Rosenthal D, Laszlo J. Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin Hematol. 1986; 23(3):177
14 - Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med.2005; 353 (1):33-45.
15 - Gisslinger H, Gotic M, Holowiecki J, et al. Final results of the ANAHYDRET-study: non-inferiority of anagrelide compared to hydroxyurea in newly diagnosed WHO-essential thrombocythemia patients. Blood. 2008; 112(11):661