Introdução:
A hiperferritinémia pode ter múltiplas causas (tabela 1), nomeadamente hereditárias (a maioria provocada por mutações no gene HFE que levam à absorção desregulada de ferro exógeno a nível do intestino delgado, sendo as mais comuns as C282Y e H63D) ou secundárias (como anemias associadas a eritropoiese ineficaz que levam a sobrecarga de ferro, administração parentérica de ferro ou doença hepática crónica). Há ainda etiologias mais raras, como é o caso da Atransferrinemia congênita (1, 2, 3).
A sobrecarga de ferro desenvolve-se de forma gradual e silenciosa, com frequência associada a sintomas inespecíficos. A deposição cumulativa de ferro em múltiplos órgãos, nomeadamente vitais, como o fígado, coração e pâncreas, pode causar falência orgânica, podendo ameaçar a vida do doente. Trata-se de um problema sério a ter em consideração quando se verifica hiperferritinémia (≥ 200ug/L nas mulheres ou ≥ 300ug/L nos homens) ou saturação de transferrina > 45%. Deve ser realizada investigação complementar da sobrecarga de ferro com ressonância magnética nuclear hepática e cardíaca. Eventualmente pode ser realizada ressonância de outros órgãos afetados, como o pâncreas, embora não seja recomendado por rotina. A biópsia hepática está reservada para os doentes em que é necessário quantificar os depósitos de ferro e a avaliar a extensão da lesão hepática (1,2,3,4).
A maioria dos doentes necessita de tratamento que consiste em flebotomias naqueles que não apresentem anemia significativa e quelantes de ferro quando esta está presente (1,2,3,4,5).
Na ausência de fibrose hepática o prognóstico é favorável, com sobrevida normal se tratamento adequado. A mortalidade está associada principalmente a doença cardíaca, cirrose hepática e carcinoma hepatocelular (2,3).
Caso clínico:
Apresenta-se o caso de um homem de 38 anos de idade, atleta de alta competição, com antecedentes pessoais de hipercolesterolemia e rinite alérgica e antecedentes familiares de cirrose hepática e diabetes mellitus no pai, falecido por insuficiência hepática em idade jovem. Medicado habitualmente com suplementação homeopática de ácido fólico e vitaminas do completo B e ferro endovenoso (que cumpria de forma regular previamente a treino em altitude). Foi encaminhado à consulta de Medicina Interna por hiperferritinémia detetada em avaliação analítica de rotina. Não apresentava queixas sistémicas associadas, alterações da condição física, mantendo treino de alta intensidade em altitude e competição. O exame objetivo não apresentava alterações. Na consulta inicial trazia avaliação analítica de ambulatório destacando-se hemoblobina 16,8 g/dL, volume globular médio 88,3 fL, ferro sérico 155 ug/dL, transferrina 233 mg/dL, ferritina 1402,2 ug/L, velocidade de sedimentação 4 mm/1ªh, sem alterações da enzimologia hepática e da função renal. A avaliação analítica do nosso Centro Hospitalar confirmou a hiperferritinémia, apresentando hemoglobina 15,7 g/dL, sem alterações do leucograma, plaquetas 131000 /uL, ferro sérico 95 ug/dL, ferritina 938 ng/mL, capacidade total de fixação do ferro 239 ug/dL, saturação de transferrina 40%, folatos e vitamina B12 dentro da normalidade, hemoglobina glicada 6,5%, creatinina 0,81 mg/dL, colesterol total 261 mg/dL, colesterol HDL 56 mg/dL, colesterol LDL 178 mg/dL, triglicerídeos 136 mg/dL, enzimologia hepática sem alterações. Realizou ecocardiograma transtorácico que não revelou alterações da função diastólica, apresentando ventrículo esquerdo com ligeira hipertrofia concêntrica das paredes, boa função sistólica global e dilatação biauricular major, compatível com o seu condicionamento físico.
Perante hiperferritinémia isolada, era importante considerar se estaria associada apenas à administração de ferro endovenoso ou se poderíamos estar perante um caso de hemocromatose hereditária (dada a história familiar de doença hepática e pancreática - pai com cirrose hepática e diabetes mellitus, contudo sem estudo genético realizado) e, ainda, avaliar se o doente apresentava sobrecarga de ferro com lesão de órgão.
Deste modo, foi quantificada a administração de ferro: o doente teria feito até à data 6 injeções/ano com sódio ferrigluconato (62.5 mg de ferro em cada 5 ml) durante 11 anos, correspondente a 375 mg/ano de ferro, um total de 4215 mg de ferro. Este é um valor considerável, mas ainda no limite para causar hemocromatose secundária, que acontece a partir de valores cumulativos de ferro na ordem dos 5000 mg, o que corresponde a 20 transfusões de concentrado eritrocitário (1,5,6).
Prosseguiu-se com o estudo genético de hemocromatose hereditária, sendo detetada a mutação p.H63D em heterozigotia. Não se detetou a mutação p.C282Y.
Para pesquisa de sobrecarga de ferro com lesão de órgão realizou ressonância magnética nuclear hepática que revelou fígado de morfologia globosa predominantemente à custa do lobo direito hepático existindo critérios de hepatomegália, contornos hepáticos regulares e ausência de alterações estruturais focais. Verificou-se critérios de sobrecarga de ferro, nomeadamente nas sequências em T1, sobretudo em fase, com existência de sinal hepático relativamente idêntico ao dos músculos para-espinhais, aspeto associado a redução do sinal também a nível da medula óssea lombar, sobretudo no estudo fora de fase. Em T2 verificou-se isointensidade do fígado em relação aos músculos para-espinhais. Na sequência eco-gradiente/T2* houve redução de sinal hepático por comparação às estruturas musculares (Figura 1). Houve ainda redução do sinal do parênquima esplénico em relação com depósito/sobrecarga de ferro.
A quantificação dos depósitos de ferro calculada na sequência de T1 foi bastante elevada, 300 umol/g, sendo o normal 36 umol/g.
Realizou também ressonância magnética nuclear cardíaca que não revelou presença significativa de ferro miocárdico.
Após os resultados dos exames complementares, concluímos tratar-se de um caso de hemocromatose secundária consequente à suplementação de ferro endovenoso, em doente com predisposição genética para sobrecarga de ferro, conferida pela mutação p.H63D em heterozigotia.
O doente foi orientado para o Centro de Alto Rendimento para realização de prova de esforço para estudo de arritmias induzidas pelo esforço. Foi orientado para consultas de Imunohemoterapia para realização de flebotomias e Genética para rastreio do filho. Atualmente mantém seguimento em consulta de Imunohemoterapia, tendo já realizado múltiplas flebotomias. Mantém-se assintomático e retomou os treinos de alta intensidade, sem intercorrências. Mantém suspensa a suplementação com ferro oral ou endovenoso e no último estudo analítico após 15 meses de terapêutica não apresentava alterações, hemoglobina 14.6 g/dl, saturação de transferrina 11 %, ferritina 102.5 ng/ml, ferro sérico 38 ug/dl, capacidade total de fixação do ferro 349 ug/dl.
Discussão:Trata-se de um caso de hemocromatose secundária associada à administração parentérica de ferro em doses controladas, em doente com mutação p.H63D em heterozigotia no gene HFE. Este genótipo encontrado indica que o indivíduo é portador da mutação p.H63D, mas não confirma o diagnóstico de hemocromatose hereditária, diagnóstico associado à presença da mutação C282Y em homozigotia em 80-85% dos casos (3). A mutação p.H63D pode estar associada a ligeiro aumento das reservas de ferro, mas só conduz a sobrecarga significativa de ferro, com consequente toxicidade orgânica, na presença de fatores de comorbilidade como diabetes, presença concomitante de doença hepática, como hepatite ou esteatose hepática, suplementação de ferro ou consumo excessivo de bebidas alcoólicas. A frequência alélica desta mutação difere entre populações, pode atingir até 20% e tem uma probabilidade de 50% de ser transmitida à descendência. (3,4,7,8,9).
Quando iniciámos o seguimento deste doente, parecia que estávamos perante um simples caso de hiperferritinémia por administração regular de ferro endovenoso, mas com a devida investigação acabámos for diagnosticar a mutação p.H63D em heterozigotia, que associada à suplementação de ferro levou ao seu depósito hepático e esplénico. Às vezes o que parece simples, não o é!
Alertamos com este caso clínico para a importância da pesquisa desta mutação quando estamos perante um doente com os fatores de risco mencionados e hiperferritinémia ou saturação de transferrina > 45%. No caso dos atletas de alta competição sob suplementação de ferro em doses controladas, a evidência de sobrecarga de ferro, em análises de monitorização, justifica ponderar prosseguir com o estudo genético. A deposição cumulativa de ferro pode causar toxicidade orgânica importante, que se não for atempadamente diagnosticada e tratada, pode conduzir a falência de órgão com consequências drásticas.
Quadro I
Causas de hiperferritinémia
| PRIMÁRIAS (HEREDITÁRIAS) | SECUNDÁRIAS | OUTRAS CAUSAS |
| | | |
| TIPO 1 HH relacionada a mutação do gene HFE (C282Y/ C282Y; C282Y/H63D) | ANEMIAS ASSOCIADAS A ERITROPOIESE INEFICAZ, COM SOBRECARGA DE FERRO: Talassemia major, anemia sideroblástica, anemia hemolítica crónica, anemia aplástica, deficiência de piruvato cinase, anemia responsiva à piroxidina | Sobrecarga de ferro neonatal |
| TIPO 2 Hemocromatose juvenil - tipo 2A: mutação na hemojuvelina HJV - tipo 2B: mutação na hepcidina HAMP | SOBRECARGA DE FERRO PARENTÉRICA: Transfusões de sangue, ferro parentérico, hemodiálise | Aceruloplasminemia |
| TIPO 3 Hemocromatose relacionada ao receptor 2 da transferrina (TfR2) | DOENÇA HEPÁTICA CRÓNICA: VHB, VHC, doença hepática alcoólica, esteatose hepática não-alcoólica; decorrente de shunt porto-cava; porfiria cutânea tarda | Atransferrinemia congênita |
| TIPO 4 Doença ferroportina (SLC4OA1) | Síndrome dismetabólica de sobrecarga de ferro | |
Figura I
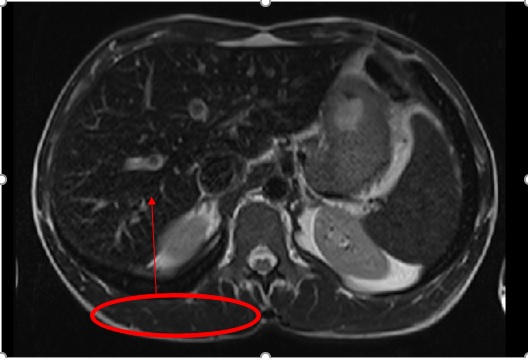
Ressonância magnética nuclear hepática - sequência eco-gradiente/T2* com redução de sinal hepático por comparação às estruturas musculares para-espinhais, em conformidade com critérios de sobrecarga de ferro
BIBLIOGRAFIA
1- Uptodate2017. Stanley L Schrier, Bruce R Bacon. Approach to the patient with suspected iron overload; [consultado 08 Setembro 2017]. Disponível em: WWW.uptodate.com.
2-B. Lorcerie, S. Audia, M. Samson, A. Millière, N. Falvo, V. Leguy-Seguin, et al. Démarche diagnostique devant une hyperferritinémie. La Revue de Médecine Interne; Aug 2015; 36 (8): 522-529.
3- Bruce R Bacon, Paul C Adams, Kris V Kowdley, Lawrie W Powell, and Anthony S Tavill. Diagnosis and Management of Hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology; Jul 2011;54(1): 328343.
4- Medscape2017. Nilesh L Vora, Ari VanderWalde. Hereditary Hemochromatosis and HFE. [consultado 08 Setembro 2017]. Disponível em: http://emedicine.medscape.com/article/1878061-overview.
5-Ángel R., Cristina S., Enric C., Cristina D.H., Joan G., et al. Guidelines on haemovigilance of post-transfusional iron overload. Blood Transfusion; Jan 2013; 11(1): 128139.
6-A. Vitor H., Ali T., Maria C. How I treat transfusional iron overload. Blood Journal 2012; 120:3657-3669.
7- Xu YY., Tang YH.,Guo XP., Wang J., Yao P. HFE genetic variability and risk of alcoholic liver disease: A meta-analysis. PubMed; Oct 2016; 36(5):626-633.
8- Pushpjeet Kanwar; Kris V Kowdley. Diagnosis and Treatment of Hereditary Hemochromatosis. Expert Rev Gastroenterol Hepatol 2013; 7(6):517-530.
9- Katrina J. Allen, Lyle C. Gurrin, Clare C. Constantine, Nicholas J. Osborne, Martin B. Delatycki, Amanda J. Nicoll,et al. Iron-OverloadRelated Disease in HFE Hereditary Hemochromatosis. N Engl J Med 2008; 358:221-230.

