

INTRODUÇÃO:
A deficiência da α-1-antitripsina (AAT) é uma doença genética, rara, que afeta o pulmão, o fígado e em alguns casos a pele. Esta surge devido a mutações na sequência de codificação do inibidor da serina protease, a AAT, que impede a sua saída do hepatócito. Como consequência existe uma diminuição da concentração de AAT na circulação predispondo ao desenvolvimento de enfisema panlobular. Além disso, devido à acumulação da glicoproteína no hepatócito vai haver inflamação hepática, fibrose e por último, cirrose.1 Estão descritas inúmeras variantes alélicas, sendo a variante M considerada normal e as variantes Z e S as que mais frequentemente se associam a doença. A variante Z quando em homozigotia associa-se a défices severos e a S tem importância clínica quando associada a outra variante patológica.2
CASO CLÍNICO:
Apresenta-se o caso de um homem, 42 anos, assintomático, sem antecedentes de relevo e sem medicação habitual, enviado à consulta externa de Medicina Interna Hepatologia, por alterações das provas de função hepática. O doente estaria a ser seguido na consulta externa de Hematologia por leucopenia e trombocitopenia.
Não existia história de hábitos tabágicos, exposição a substâncias hepatotóxicas (álcool, medicação, drogas de abuso, chás/suplementos), transfusões, antecedentes de hepatites víricas ou outras, nem história familiar relevante.
Relativamente ao exame físico salientar a presença de hepatoesplenomegalia, sem ascite. Analiticamente apresentava leucócitos 3,60 x10^9/L (4,50 - 11,50); plaquetas 56,0 x10^9/L (150,0 - 450,0); TP 14,1 segundos (9,9 - 12,8); INR 1,19; fosfatase alcalina (FA) 76 UI/L (25 - 100U); GGT 130 UI/L (7 - 49); ALT 50 UI/L (4 - 43); AST 46 UI/L (4 - 43); bilirrubina total 1,6 mg/dL (0,3 - 1,2); bilirrubina direta 0,54 mg/dL (0,10 - 0,50); albumina 4 g/dL (3,5 - 5,0); α-1-antitripsina 24,60 mg/dL (80 - 199) e apresentava ainda hipergamaglobulinemia de base alargada, compatível com doença hepática crónica. O restante estudo analítico complementar auto-imune (ANA, ASMA e AMA) e serológico - HBV, HCV, HIV, CMV e EBV foi negativo. Neste contexto foi pedida ecografia abdominal e fenotipagem de α-1-antitripsina (focagem isoeléctrica) que revelaram marcada esplenomegalia com aparente hipertrofia do lobo esquerdo do fígado e fenótipo MS.
Uma vez que, de acordo com a literatura existente, este não se encontrava associado a doença prossegui-se com o estudo. A endoscopia digestiva alta mostrou varizes esofágicas médias e eritema de congestão difusa da mucosa gástrica, compatível com gastropatia de hipertensão portal moderada. Foi feita TC-abdominal que evidenciou fígado com hipertrofia relativa do lobo esquerdo de contornos ondulados e textura discretamente heterogénea, de acordo com hepatopatia crónica, sem imagens nodulares. A veia porta tinha calibre aumentado. Verificavam-se ainda múltiplas varizes esofágicas, do fundo gástrico e do hilo esplénico, bem como repermeabilização da veia para-umbilical, associadas a marcada esplenomegalia (20 cm) homogénea. Sem líquido livre intra-abdominal. Por fim, o doente realizou biópsia hepática transjugular que revelou alguns corpos de Mallory periseptais, esteatose microvacuolar focal moderada, sendo que focalmente existiam grânulos intracitoplasmáticos PAS +, diastase resistentes que expressam AAT, compatíveis com o diagnóstico de deficiência de AAT. Havia fibrose moderada com septos porto-portais. (Fig. 1) Perante este diagnóstico completou-se o estudo com TC-tórax e provas de função respiratória sem alterações de relevo. O doente manteve seguimento em consulta de pré-transplante, tendo sido submetido recentemente a transplante hepático.
DISCUSSÃO:
A suspeição clínica desta entidade deve surgir em doentes que apresentem enfisema em idades jovens, predominantemente nas bases pulmonares e sem história de tabagismo, naqueles com história familiar de enfisema e/ou doença hepática crónica , com história clínica ou sinais de paniculite, doença hepática crónica de etiologia não esclarecida ou mesmo em doentes assintomáticos com padrão obstrutivo persistente em provas de função respiratória.3 As manifestações clínicas são várias e dependem da lesão associada: enfisema, cirrose, paniculite ou vasculite. Nos adultos com deficiência de AAT a doença pode surgir apenas com alteração das provas de função hepática, em doentes assintomáticos, como é o caso deste doente, com manifestações clínicas de cirrose avançada ou mesmo carcinoma hepatocelular.1
Existem inúmeras variantes alélicas, como já foi referido, que estão associadas a diferentes fenótipos. Resumidamente existem 4 grupos: normal, associado a níveis normais de AAT; o deficiente, com níveis <35% dos valores normais de AAT; o nulo, onde a AAT não é detetada em circulação e o disfuncional, com produção normal da proteína, mas a mesma não funciona apropriadamente.
A doença surge com mutações no produto genético normal, PiM, dando origem às variantes mais comuns PiS, que expressa cerca de 50-60% de AAT, especialmente prevalente no sul da Europa, nomeadamente em Portugal e PiZ, que expressa cerca de 10-20%, prevalente em países do norte da Europa.4 Os fenótipos mais frequentemente encontrados são o PiMS, com uma prevalência que varia entre 4-11% na Europa 2, 5 e o PiMZ. Dos que estão associados a deficiência o PiSS, o PISZ e o PiZZ são os mais frequentes. Outros, mais raros, mas que estão associados a doença hepática são o Mmalton e o Mduarte.6 Os doentes heterozigotos, MZ, cerca de 2-3% da população, têm níveis de AAT entre 50-70% do normal e possuem um risco ligeiramente aumentado de desenvolver DPOC 7 e poderão ter um maior risco de vir a desenvolver doença hepática.8, 9 O alelo S quer em homozigotia quer associado ao M, não está associado a doença hepática.1 Contudo quando associado ao alelo Z existe uma franca predisposição a desenvolver doença hepática 1, 9-11 e enfisema, apesar de apresentar menor risco que o fenótipo ZZ.2 Os dois alelos que estão descritos como estando mais frequentemente associados a doença hepática são o Z e Mmalton. Existem outros descritos, que estão associados à acumulação, dentro dos hepatócitos, da AAT como o Mduarte, Mnichinan, S e Siiyama.7, 12-14
O caso aqui descrito diz respeito a um doente assintomático que se apresenta com alterações da enzimiologia hepática associadas a sinais indiretos de cirrose. Assim sendo as hipóteses de diagnóstico mais comuns são: doença hepática alcoólica ou não alcoólica, hepatite B ou C crónica. Outras causas menos comuns são: hepatite auto-imune, colangite esclerosante primária, colangite biliar primária e doenças metabólicas hereditárias como a hemocromatose, doença de wilson e deficiência da alfa 1 anti-tripsina. A doença hepática alcoólica seria improvável dado não existir história de abuso de álcool; a doença hepática não alcoólica, encontra-se habitualmente associada a doentes com obesidade e história de diabetes mellitus em contexto de síndroma metabólica, o que não se verificava neste caso. Relativamente às infeções víricas, estas foram excluídas após serologia negativa.
Após constatação de valores baixos de AAT foi pedida a fenotipagem, sendo que o restante estudo analítico inicial foi negativo. Uma vez que o fenótipo, de acordo com a literatura não está associado a doença foi colocada a hipótese de esta entidade não ser a causa, sendo que se optou por proceder com o estudo, tendo-se optado por realizar biópsia hepática que confirmou o diagnóstico e excluíu outras causas que poderiam estar na origem da evolução cirrogénica. Deste modo não foi pedida genotipagem. Existe a possibilidade de algum fator genético e/ou adquirido, não identificado até à data, ter contribuído para o desenvolvimento da mesma.
CONCLUSÃO:
A relevância deste caso surge com o fato de um fenótipo, que está associado a cerca de 80% dos níveis normais de AAT e como tal, até à data, conhecido por não ser causador de doença poder na realidade ser responsável por doença hepática avançada.
Figura I
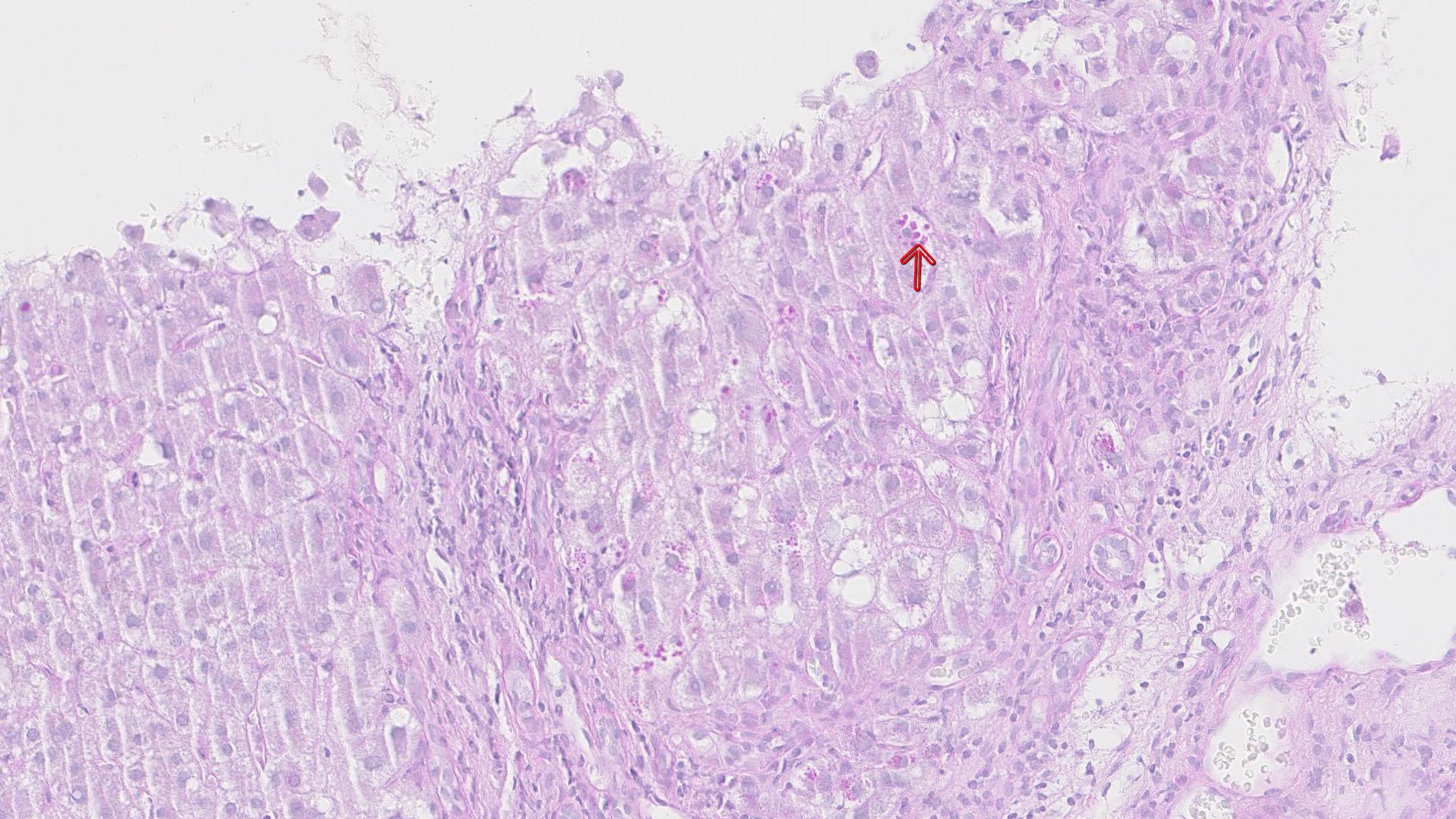
Hepatócitos com glóbulos citoplasmáticos PAS + diastase resistentes com distorção fibrótica arquitetural sugestiva de evolução cirrogénica
BIBLIOGRAFIA
1. Fairbanks KD, Tavill AS. Liver disease in alpha 1-antitrypsin deficiency: a review. The American journal of gastroenterology. 2008;103(8):2136-41; quiz 42.
2. Abboud RT, Nelson TN, Jung B, Mattman A. Alpha1-antitrypsin deficiency: a clinical-genetic overview. The application of clinical genetics. 2011;4:55-65.
3. Hogarth DK, Rachelefsky G. Screening and familial testing of patients for alpha 1-antitrypsin deficiency. Chest. 2008;133(4):981-8.
4.de Serres FJ, Blanco I, Fernández-Bustillo E. Genetic epidemiology of alpha-1 antitrypsin deficiency in North America and Australia/New Zealand: Australia, Canada, New Zealand and the United States of America. Clin Genet. 2003;64(5):382-97
5. Hutchison DC. Alpha 1-antitrypsin deficiency in Europe: geographical distribution of Pi types S and Z. Respiratory medicine. 1998;92(3):367-77.
6. Cox DW, Billingsley GD. Rare deficiency types of alpha 1-antitrypsin: electrophoretic variation and DNA haplotypes. American journal of human genetics. 1989;44(6):844-54.
7. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. American journal of respiratory and critical care medicine. 2003;168(7):818-900.
8. Graziadei IW, Joseph JJ, Wiesner RH, Therneau TM, Batts KP, Porayko MK. Increased risk of chronic liver failure in adults with heterozygous alpha1-antitrypsin deficiency. Hepatology. 1998;28(4):1058-63.
9. Teckman JH, Qu D, Perlmutter DH. Molecular pathogenesis of liver disease in alpha1-antitrypsin deficiency. Hepatology. 1996;24(6):1504-16.
10. Mahadeva R, Chang WS, Dafforn TR, Oakley DJ, Foreman RC, Calvin J, et al. Heteropolymerization of S, I, and Z alpha1-antitrypsin and liver cirrhosis. The Journal of clinical investigation. 1999;103(7):999-1006.
11. Lomas DA, Mahadeva R. Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. The Journal of clinical investigation. 2002;110(11):1585-90.
12. Salahuddin P. Genetic variants of alpha1-antitrypsin. Current protein & peptide science. 2010;11(2):101-17.
13. Seyama K, Nukiwa T, Takabe K, Takahashi H, Miyake K, Kira S. Siiyama (serine 53 (TCC) to phenylalanine 53 (TTC)). A new alpha 1-antitrypsin-deficient variant with mutation on a predicted conserved residue of the serpin backbone. The Journal of biological chemistry. 1991;266(19):12627-32.
14. Janciauskiene S, Eriksson S, Callea F, Mallya M, Zhou A, Seyama K, et al. Differential detection of PAS-positive inclusions formed by the Z, Siiyama, and Mmalton variants of alpha1-antitrypsin. Hepatology. 2004;40(5):1203-10.