Introdução
A Síndrome de Sneddon (SS), descrita pela primeira vez em 1965, é definida pela associação de acidente vascular cerebral (AVC) e livedo reticular (LR), com achados histopatológicos característicos na biopsia cutânea (trombose das arteríolas subcutâneas, dilatação capilar compensatória e estase sanguínea).1 Acidentes isquémicos transitórios e a existência de pequenos focos hiperintensos na ponderação T2 da ressonância magnética (RM) cerebral constituem critérios de suporte.2 Tem uma incidência de 4/1000000/ano, estando presente em até 0,5% dos doentes internados por AVC.3 É mais comum em mulheres em idade fértil, não apresenta diferenças étnicas e ocorre maioritariamente de forma esporádica.3,4 Subsistem dúvidas quanto à sua fisiopatologia. Menos de 50% dos casos associam-se à Síndrome Antifosfolípido (SAF).3 Apresenta-se um caso de SS associado a SAF, com o intuito de alertar para esta síndrome subdiagnosticada.
Caso Clínico
Mulher de 46 anos, caucasiana, cozinheira, tendo como principais problemas hipertensão arterial (bem controlada com lisinopril+hidroclorotiazida 20+12,5mg/dia), tabagismo (carga 16 unidades maço-ano), depressão reativa (tratada com mirtazapina 15mg/dia) e enxaquecas frequentes desde a infância. História de menarca aos 14 anos, duas gravidezes bem sucedidas com necessidade de cesariana por não progressão do feto no canal de parto e laqueação tubar aos 30 anos. Sem história de fenómenos trombóticos. Antecedentes familiares irrelevantes.
A doente foi submetida a colecistectomia laparoscópica programada por colecistite crónica sintomática, tendo tido alta no próprio dia e regressado ao trabalho em menos de uma semana, sem evidência de complicações imediatas. Um mês depois, desenvolveu subitamente desvio da comissura labial para a esquerda com duração de minutos e desde então persistiu défice da memória de trabalho.
Recorreu ao Médico de Família, tendo realizado tomografia cerebral, que mostrou hipodensidade cortico-subcortical temporo-insular esquerda com extensão à substância branca da coroa radiada e outras de menores dimensões na região frontoparietal bilateralmente, sugestivas de lesões vasculares isquémicas sequelares. Iniciou Clopidogrel 75mg/dia e foi enviada a consulta de Neurologia e Medicina Interna. Apresentava linguagem pouco fluente, sem alteração da nomeação, repetição e compreensão. Mini-Mental State Examination (6º ano de escolaridade) de 23/30 correspondendo a défice cognitivo ligeiro. Sem alterações no restante exame neurológico. A pele apresentava padrão de descoloração cianótica de aspeto rendilhado nas nádegas, coxas e joelhos, sugestivo de LR (Fig. 1), que a doente já apresentaria há vários anos, de forma fixa e independente da temperatura ou posição do corpo.
A ressonância magnética cerebral mostrou alargamento dos espaços de circulação do líquor e confirmou sequelas de enfartes cortico-subcorticais bilaterais (Fig. 2). Ecodoppler carotídeo e vertebral, ecocardiograma e Holter não mostraram alterações. Laboratorialmente, destaca-se tempo de tromboplastina parcial ativada (APTT) aumentado (96,7 s), anticorpos anti-nucleares (ANA) positivos (título 1:1280) e anticoagulante lúpico (> 2,06), anticorpos anti-cardiolipina (IgG >120 GPL, IgM >46,6 MPL) e anti-beta-2-glicoproteína (IgG >37,5 U/mL, IgM >44,1 U/mL) positivos em mais que 2 ocasiões (mais de 12 semanas de intervalo). A velocidade de sedimentação (VS) era discretamente elevada (36mm/1ªh), sem elevação de outros marcadores laboratoriais de inflamação e sem citopenias. Restante estudo imunológico, que incluía anti-dsDNA e ENA, foi negativo.
Foi feito o diagnóstico de SAF em mulher jovem, com fatores de risco vascular, doença cerebrovascular isquémica bilateral e presença de anticorpos antifosfolípido. A presença adicional de LR configura a SS.
Iniciou hipocoagulação com acenocumarol em Novembro de 2014 e manteve antiagregação com clopidogrel. Contra o conselho médico, não conseguiu deixar de fumar.
Verificou-se melhoria franca das cefaleias e diminuição da extensão e intensidade do LR nos meses que se seguiram ao início da hipocoagulação. Porém existe noção subjectiva de que o défice de memória continuou a progredir nos anos que se seguiram, apesar da ausência de progressão imagiológica. Não se verificaram novos eventos trombóticos ou outras manifestações de SAF.
Discussão
A fisiopatologia da Síndrome de Sneddon não é totalmente compreendida. A evidência de trombose vascular e recanalização na pele e no cérebro bem como várias alterações analíticas relacionadas com a coagulação suportam a convicção da coagulopatia como principal mecanismo. Está descrito que até 50% dos casos se associa à SAF3 mas se além de se pesquisarem anticorpos anti-cardiolipina, anti-beta-2-glicoproteína e o anticoagulante lúpico forem também avaliados outros anticorpos anti-fosfolípido (AFL), a associação entre SS e SAF é ainda maior. Dutra et al mostrou que quando se pesquisam também os anticorpos anti-protrombina e anti-fosfatidiletanolamina, até 80% dos doentes com SS apresenta pelo menos um destes cinco positivo.5 Além disso, o facto dos títulos destes anticorpos serem flutuantes ao longo do tempo e de nem sempre serem medidos através dos métodos laboratoriais definidos internacionalmente, fazem com que a percentagem de doentes com critérios analíticos de SAF possa ser muito variável.6 Outras coagulopatias descritas na SS são a resistência à proteína C ativada, deficiência de antitrombina ou das proteínas S ou Z, alterações do rácio ativador/inibidor do plasminogénio e defeitos da agregação plaquetária.3 Está descrita uma forma familiar da SS associada a uma mutação no gene CECR1 que resulta no défice de ADA2.7 Existem outras etiologias descritas e que incluem como mecanismos arteriopatia primária de pequenos/médios vasos ou disfunção endotelial secundária a doenças autoimunes. 3,8 A SS pode associar-se ou fazer parte do espetro de algumas doenças, como SAF e Lúpus Eritematoso Sistémico, por partilharem várias características clínicas como LR, enxaqueca, AVC, alterações cognitivas e laboratoriais.3,5
O LR consiste num padrão cutâneo rendilhado de descoloração eritemato-cianótica. Alguns autores distinguem duas formas: 1) livedo reticular se a trama reticulada ocorre de forma completa com interligação nítida e 2) livedo racemosa quando esta trama é sucessivamente interrompida. O segundo é com maior frequência patológico e mais comum na SS. Na SS, o LR localiza-se habitualmente nas pernas (100%), tronco (84-89%) e nádegas (68-74%), persiste com o calor e é a manifestação inicial em 17,5%.3 É mais frequente na SS com anticorpos AFL negativos (89% vs 21%, p<0.001).9 Também está presente em 16% dos doentes com SAF e nestes existe uma forte associação entre o LR e eventos trombóticos arteriais (OR 10,8), sendo considerado um fator importante na decisão terapêutica.10,11
A SS está presente em até 0,5% dos doentes internados por AVC, sendo importante considerá-la particularmente no jovem.12 As lesões cerebrais são frequentemente pequenas e multifocais e predominam na substância branca profunda periventricular.3,12,13,14
As cefaleias são o sintoma mais frequente da SS, apesar de inespecíficas. Metade dos doentes tem enxaquecas, sendo mais frequentes nos doentes com anticorpos AFL.3,13 As alterações cognitivas (défice de concentração, atenção, memória, perceção visual e construção visuoespacial) e psiquiátricas (depressão e demência precoce) estão presentes em 77%.3,14 Na SS, fruto do envolvimento preferencial de pequenos/médios vasos, estas alterações são progressivas ao longo dos anos e resultam do efeito cumulativo de múltiplos AVC, por vezes subclínicos.3,13,14 Na SAF os défices neurológicos são mais estáveis, uma vez que os vasos afetados são habitualmente de maior calibre.15
Outras manifestações descritas incluem a HTA, doença cardíaca valvular e isquémica, disfunção renal progressiva, epilepsia e alterações oftalmológicas.3
Não existem ainda estudos de qualidade que permitam definir o tratamento ideal da SS. Tendo por base a sua provável patogénese e sobretudo quando se associam a anticorpos AFL positivos ou anticoagulante lúpico é recomendado o início de hipocoagulação com varfarina, acenocumarol ou heparina de baixo peso molecular.3 Nestes casos, as recomendações são semelhantes às da SAF. Um estudo mostrou o benefício do ácido acetilsalicílico na SS com anticorpos AFL negativos, mas não da hipocoagulação.9 A nifedipina parece reduzir as manifestações cutâneas, porém não previne as complicações cerebrovasculares.3 Os corticóides, a ciclofosfamida e a azatioprina parecem ser ineficazes.3
Descreveu-se o caso de uma mulher fumadora com enxaqueca e LR e evidência clínica e imagiológica de múltiplas lesões isquémicas cerebrais, configurando a SS. A investigação levou ao diagnóstico adicional de SAF. Levanta-se a possibilidade da cirurgia ter sido o second hit numa doente com anticorpos AFL positivos. Mais característico da SS do que da SAF foi a evolução lenta e progressiva do défice cognitivo, bem como a distribuição das lesões cerebrais. Destaca-se a melhoria do LR e das cefaleias após o início da hipocoagulação. A opção de manter antiagregação deveu-se à presença de outros fatores de risco vascular e de lesões na substância branca.
A descrição deste caso visa alertar para a existência de sinais como o LR que podem constituir a pista para a etiologia do AVC no jovem. É essencial uma história clínica detalhada que inclua a pesquisa de sintomas como enxaqueca, a história obstétrica e cirurgias recentes, bem como um exame físico exaustivo com pesquisa de LR nas localizações típicas. Além da revisão dos fatores de risco vascular clássicos, sugerimos a pesquisa sistemática dos anticorpos AFL e anticoagulante lúpico nos doentes jovens com AVC, dadas as implicações terapêuticas. Continua a não ser clara a terapêutica nas situações de SS com AFL e anticoagulante lúpico negativos. A decisão deve ser ponderada caso a caso e parece lícito tentar anti-agregante plaquetário e hipocoagulação caso o primeiro não seja bem sucedido. Dada a sua ausência, os autores reconhecem a importância da realização de estudos epidemiológicos na população portuguesa de modo a avaliar a prevalência da síndrome de Sneddon e da SAF nos doentes com AVC.
Figura I
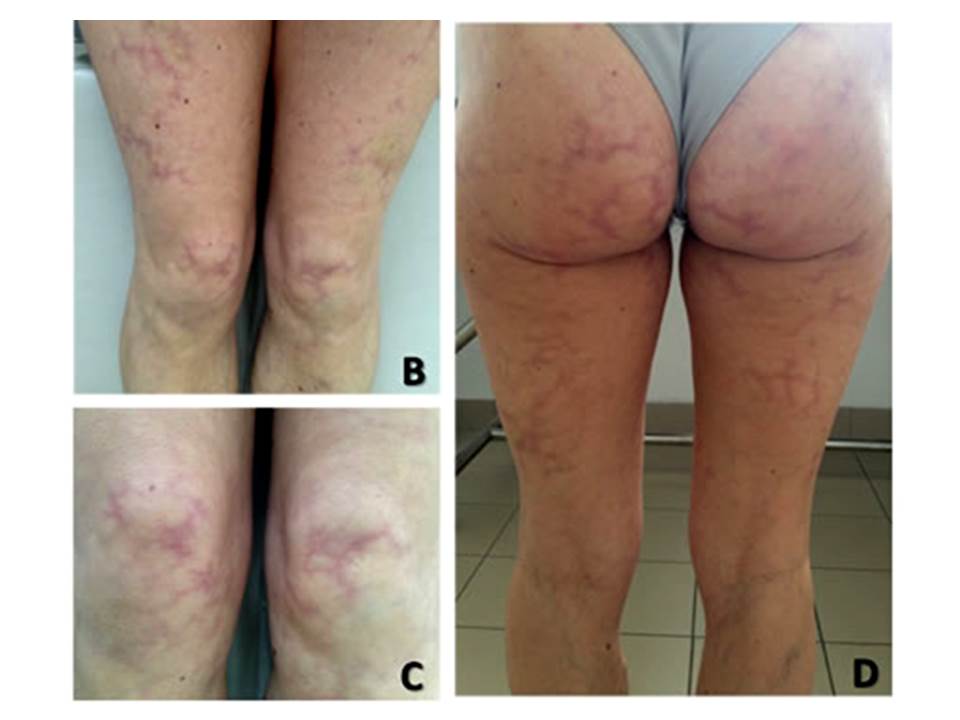
Figura 1 Livedo racemosa nas coxas (A e C), joelhos (A e B) e nádegas (C).
Figura II
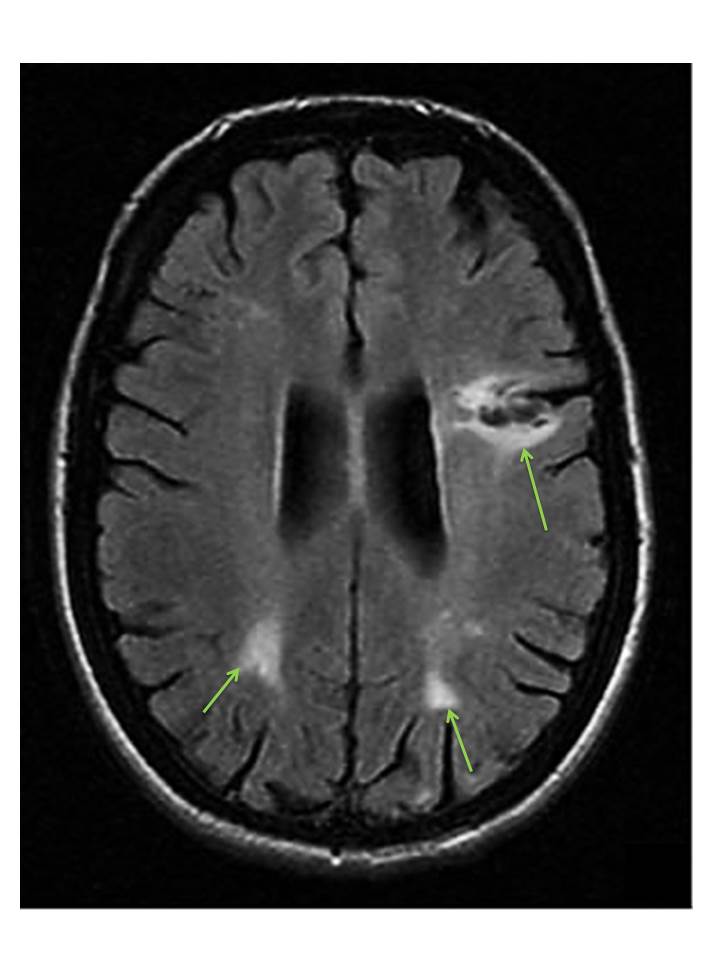
Figura 2 Sequelas de enfartes cortico-subcorticais bilaterais (setas) e alargamento dos espaços de circulação do líquor, em RM.
BIBLIOGRAFIA
1. Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180185.
2. Schellong SM, Weissenborn K, Niedermeyer J, Wollenhaupt J, Sosada M, Ehrenheim C, et al. Classification of Sneddons syndrome. Vasa. 1997;26:215221.
3. Wu S, Xu Z, Liang H. Sneddons syndrome: a comprehensive review of the literature. OrphanetJRareDis. 2014;9(215):1-7.
4. Aladdin Y, Hamadeh M, Butcher K. The Sneddon syndrome. Arch Neurol. 2008;65(6):834-835.
5. Dutra LA, Braga-Neto P, Pedroso JL, Barsottini O. Sneddons syndrome: case report and review of its relationship with antiphospholipid syndrome. Einstein. 2012;10(2):230-232.
6. France`s C, Piette J. The Mystery of Sneddon Syndrome: Relationship with Antiphospholipid Syndrome and Systemic Lupus Erythematosus. J Autoimmun. 2000; 15, 139-143.
7. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370:911-920.
8. Cirillo G, Tessitore A, Cirillo M, Salemi F, Liguori S, Esposito S, et al. Livedo and ischemic strokes: diagnostic hints of a rare condition. Neurol Sci. 2013;34:2073-2075.
9. Francès C, Papo T, Wechsler B, Laporte JL, Biousse V, Piette JC. Sneddon syndrome with or without antiphospholipid antibodies. A comparative study in 46 patients. Medicine (Baltimore). 1999;78(4):209-19.
10. Abreu MM, Danowski A, Wahl DG, Amigo MC, Tektonidou M, Pacheco MS, et al. The relevance of ´non-criteria´ clinical manifestations of antiphospholipid syndrome: 14th International Congress on Antiphospholipid Antibodies Technical Task Force Report on Antiphospholipid Syndrome Clinical Features. Autoimmun Rev. 2015;14(5):401-14.
11. Erkan D, Lockshin MD. Non-criteria manifestations of antiphospholipid syndrome. Lupus. 2010;19(4):424-7.
12. Boesch SM, Plörer AL, Auer AJ, Poewe W, Aichner FT, Felber SR, et al. The natural course of Sneddon syndrome: clinical and magnetic resonance imaging findings in a prospective six year observation study. J Neurol Neurosurg Psychiatry. 2003;74:542544.
13. Tietjen GE, Al-Qasmi MM, Gunda P, Herial NA. Sneddons syndrome: another migraine-stroke association? Cephalalgia. 2006;26:225232.
14. Adair JC, Digre KB, Swanda RM, Hartshorne MF, Lee RR, Constantino TM, et al. Sneddons syndrome: cause of cognitive decline in young adults. Neuro Psychiatry Neuropsychol Behav Neurol. 2001;14:197204.
15. Marinho J, Piovesan EJ, Neto M, Kotze LR, de Noronha L, Twardowschy CA, et al. Clinical, neurovascular and neuropathological features in Sneddon ́s Syndrome. Arq Neuropsiquiatr. 2007;65(2):390-395.

