INTRODUÇÃO
Existem numerosas causas de doença hepática que resultam em cirrose, causando inflamação hepática cronica ou colestase. O consumo de álcool, infeção pelo vírus da hepatite B e C, e síndrome metabólica, devido ao excesso de peso e obesidade, são as principais causas de cirrose e hepatocarcinoma na Europa. 1
Alfa-1 antitripsina (A1AT) é uma glicoproteína inibidora da enzima elastase neutrofílica, produzida no fígado, cuja principal função é hidrolisar as fibras de elastina no pulmão. O gene da A1AT é polimórfico e por isso apresenta vários fenótipos (Z, S, M e null) que diferem na quantidade e qualidade de proteína normal produzida. Esta variabilidade traduz-se clinicamente em espectros muito distintos de gravidade de doença e idade da sua manifestação, sendo o atingimento predominante do pulmão (enfisema e bronquiectasias) secundária à deficiência desta enzima, e do fígado (hepatite colestática e cirrose) secundária a acumulação da enzima com configuração anormal em forma de corpúsculos de inclusão nos hepatócitos. O diagnóstico baseia-se em baixos níveis séricos de A1AT, associados a um fenótipo deficitário. Aproximadamente 10 a 15% dos adultos com défice de A1AT desenvolvem doença hepática.2 Cerca de 40% dos adultos com o fenótipo PI*ZZ, apresentam histologicamente doença hepática importante e cirrose. 3
Não existe, atualmente, nenhum tratamento dirigido para a doença hepática, uma vez que o mecanismo de lesão se deve à acumulação da proteína Z mutada, e não à deficiência da protesase. 3
O transplante pulmonar e/ou hepático (de acordo com o principal órgão atingido) é o único tratamento com impacto na mortalidade. 4 O défice de A1AT pode também associar-se a glomerulonefrite, do tipo membranoproliferativa, em associação ao fenótipo PI*ZZ, ou nefropatia por IgA, associada a cirrose hepática. 5
CASO CLÍNICO
Doente do sexo masculino, 52 anos de idade, caucasiano. Recorreu ao serviço de urgência em Janeiro de 2014, por quadro de astenia e anorexia, associados a edemas dos membros inferiores e aumento do perímetro abdominal com 3 meses de evolução. Referia também dispneia para médios esforços e ortopneia. Noção de diminuição do débito urinário e ocasionalmente com hematúria vestigial. Negava emagrecimento, febre, náuseas, vómitos, dor abdominal, disúria ou polaquiúria, alterações do trânsito gastrointestinal.
Dos antecedentes pessoais, a salientar: hipertensão arterial essencial, doença hepática conhecida desde 2011, sem estudo etiológico, mas atribuída ao consumo de álcool (cerca de 250g/dia), segundo o doente abstinente há 1 ano. Endoscopia digestiva alta recente sem varizes esofágicas. Doença renal crónica estadio 3 de etiologia não esclarecida, apresentando hematúria macroscópica intermitente desde 2011. Anemia normocítica e normocrómica, hipoproliferativa e sem défices vitamínicos associados. Dislipidemia e não fumador. Residente em ambiente rural, com animais domésticos (porcos, galinhas e coelhos). Sem saneamento básico e com consumo de água de poço (sem análise de qualidade regular).
No exame objetivo à admissão no serviço de urgência apresentava razoável estado geral, discurso lentificado e flapping. Mucosas descoradas e hidratadas, anictérico. Sem turgescência venosa jugular ou refluxo hepato-jugular a 45º. Sem sinais de dificuldade respiratória. Sem alterações à auscultação cardíaca ou pulmonar. Evidência de circulação colateral torácica e abdominal. Abdómen com ascite de grande volume, sem tensão, mole e depressível, indolor, sem sinais de irritação peritoneal, hepatomegalia ligeira com bordos lisos e indolor. Edemas dos membros inferiores até aos joelhos e na face posterior das coxas. Algaliado, com saída de urina hemática.
Laboratorialmente constatado agravamento da anemia (hemoglobina 6.2g/dL) normocítica e normocrómica, trombocitopenia (36x109/L), agravamento da função renal (creatinina sérica 7.7mg/dL e ureia sérica 181mg/dL) com acidemia metabólica e hipercalémia. Transaminases normais. Hipoalbuminemia. Estudo da coagulação com prolongamento do tempo de tromboplastina parcial ativada e de protrombina. O sedimento urinário demonstrava eritrocitúria e proteinúria (1g/L). Ecografia reno-vesical excluiu componente obstrutivo.
Realizada paracentese evacuadora, sendo o gradiente albuminocitogénico do líquido ascítico aumentado, sem critérios de peritonite bacteriana espontânea.
Assumida doença renal crónica agudizada com necessidade de hemodiálise e cirrose hepática com hipertensão portal tendo ficado internado no serviço de Medicina Interna. Do estudo etiológico da doença hepática: antigénio e anticorpo-HIV1 e 2, anticorpo-HCV, antigénio HBs, anticorpo-HB e anticorpo-HBs negativos. Níveis séricos de alfa-1 anti-tripsina reduzidos 22.4mg/dL (normal 103-302mg/dL). Imunoglobulina G e A aumentadas. Cinética do ferro e cobre normais. Estudo auto-imune sem alterações. Proteinúria nefrítica (1g/L) e eritrocitúria. Realizou ecografia abdominal, que demonstrou fígado com estigmas de hepatopatia cronica, com contornos lobulados e estrutura heterogénea, assim como ligeira esplenomegalia homogénea e ascite de moderado volume.
A biópsia hepática transjugular demonstrou cirrose hepática com acumulação de glóbulos hialinos PAS e PAS-D resistentes em hepatócitos - alterações morfológicas sugestivas de etiologia tóxica/alcoólica, sendo positiva a pesquisa de glóbulos de alfa-1 antitripsina por estudo imunohistoquímico. (Fig. 1)
Realizou ainda biópsia renal que documentou glomerulonefrite mesangioproliferativa e necrose tubular aguda. (Fig. 2) Ao longo do internamento, o doente apresentou recuperação da função renal sem necessidade de manutenção de hemodiálise. Manteve ascite refratária, com necessidade de paracenteses regulares.
Para estratificação da doença associada a deficiência A1AT, realizou TC-torácica que documentou enfisema nos lobos inferiores (característica desta patologia) e provas funcionais respiratórias compatíveis com síndrome ventilatório obstrutivo moderado, tendo iniciado terapêutica broncodilatadora. Realizado ainda estudo genético da A1AT que comprovou fenótipo PI-ZZ.
Por se tratar de doente com cirrose hepática [Child-Pugh C (10 pontos) e MELD 23] de etiologia alcoólica associada a deficiência de A1AT, e por se encontrar abstinente há mais de um ano, foi proposto para duplo transplante (fígado e rim).
Após o internamento, o doente manteve necessidade de paracenteses evacuadoras 1 vez por mês e sem necessidade de HD até Novembro desse ano. Em Fevereiro de 2015 foi admitido no nosso hospital com quadro de insuficiência hepática acabando por falecer antes da transplantação.
DISCUSSÃO
Se a doença pulmonar em idade jovem é frequentemente causa de pesquisa desta mutação, a cirrose hepática no adulto (que surge em doentes com manifestações pulmonares menos graves) é muitas vezes esquecida e por isso subdiagnosticada.
A doença hepática apenas foi observada em indivíduos portadores de alelos que originem produção da proteína (Z e M), não sendo por isso observada nos PI*NN, uma vez que não há proteína circulante, não ocorrendo consequente acumulação no hepatócito, mecanismo responsável pela lesão hepática. 6 Nos restantes fenótipos, aproximadamente 10 a 15% dos adultos desenvolvem doença hepática. 2
O consumo excessivo de álcool está associado a uma variedade de alterações hepáticas, incluindo hepatite alcoólica e cirrose hepática. Doentes com consumo de álcool superior a 30g por dia apresentam maior risco de cirrose, apesar da maioria não a desenvolver apesar de consumos alcoólicos excessivos. 7
Perante o caso clínico apresentado, apesar do consumo alcoólico excessivo que o doente apresentara no passado, mantinha-se a dúvida da co-existência de outra etiologia. Excluídas outras possíveis causas (doença de Wilson, hemocromatose, hepatite vírica) e dado o défice exuberante da proteína A1AT, pareceu-nos perentória a realização de biópsia hepática, cujos achados confirmaram uma etiologia mista - défice de A1AT sobreposta a álcool. Sendo o genótipo do doente compatível com manifestação fenotípica, a pesquisa de doença pulmonar é também fundamental. De relembrar que o doente não tinha história tabágica nem exposição profissional que justificassem as alterações pulmonares identificadas, pelo que estas serão atribuíveis ao défice da proteína A1AT.
Quanto à etiologia da doença renal crónica, apesar da suspeita inicial de nefropatia por deposição de IgA, habitualmente associada a doença hepática crónica, esta não foi confirmada, tendo-se identificado uma forma de doença glomerular que não se encontra descrita como associada a depósito de A1AT.
Não existe atualmente nenhum tratamento específico para a doença hepática, uma vez que a lesão hepática ocorre por deposição de proteína com configuração anormal e não pelo seu défice. Preconizam-se medidas para prevenir e reduzir as complicações da doença hepática cronica. O transplante hepático está indicado nos doentes com doença hepática terminal, resultando numa sobrevivência de 85% aos 5 anos.4 O doente em causa foi proposto para duplo transplante (fígado e rim) dado o estadio avançado da doença hepática e renal, no entanto devido a falta de condições socioeconómicas, com consequente dúvida de cumprimento do seguimento posterior necessário, não foi inserido em lista de transplante, que seria o único tratamento curativo que lhe poderíamos oferecer, acabando por falecer de insuficiência hepática.
Os autores querem com este caso relembrar a deficiência de A1AT como uma possível causa de cirrose hepática no adulto, cujo diagnóstico tem implicações terapêuticas, não apenas no que diz respeito à doença hepática, mas pela associação a doença respiratória, muitas vezes pauci-sintomática.
AGRADECIMENTOS
Um especial agradecimento ao Dr. Pedro Rodrigues Pereira, à Dra. Joanne Lopes e à Dra. Laura Pontes Fajardo, do serviço de Anatomia Patológica do Hospital de São João, por gentilmente cederem as imagens das biópsias renal e hepática.
Figura I
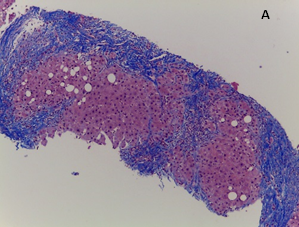
Figura 1: Biópsia hepática: A Fígado em estadio de cirrose; B Glóbulos hialinos intracitoplasmáticos PAS e PAS-D positivos, com pesquisa de alfa-1 antitripsina positiva por estudo imunohistoquímico (seta).
Figura I
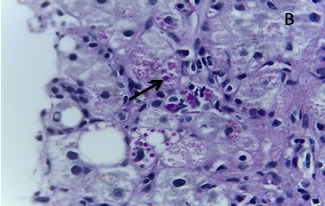
Figura 1: Biópsia hepática: A Fígado em estadio de cirrose; B Glóbulos hialinos intracitoplasmáticos PAS e PAS-D positivos, com pesquisa de alfa-1 antitripsina positiva por estudo imunohistoquímico (seta).
Figura II
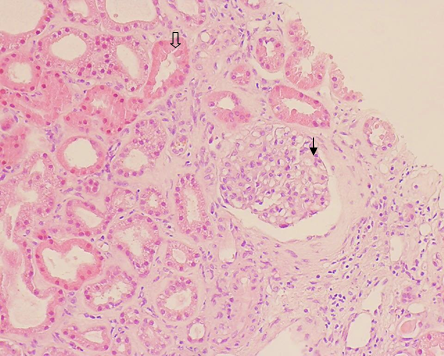
Fig. 2 - Biópsia Renal zonas de necrose tubular aguda (seta vazia) e proliferação mesangial (seta preenchida).
BIBLIOGRAFIA
1. Blachier M, Leleu H, Peck-Radosavljevic M, Valla DC, Roudot-Thoraval F. The burden of liver disease in Europe: a review of available epidemiological data. Journal of hepatology. 2013 Mar;58(3):593-608. PubMed PMID: 23419824.
2. American Thoracic S, European Respiratory S. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. American journal of respiratory and critical care medicine. 2003 Oct 01;168(7):818-900. PubMed PMID: 14522813.
3. Bals R. Alpha-1-antitrypsin deficiency. Best practice & research Clinical gastroenterology. 2010 Oct;24(5):629-33. PubMed PMID: 20955965.
4. Carey EJ, Iyer VN, Nelson DR, Nguyen JH, Krowka MJ. Outcomes for recipients of liver transplantation for alpha-1-antitrypsin deficiency-related cirrhosis. Liver transplantation : official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2013 Dec;19(12):1370-6. PubMed PMID: 24019185.
5. Davis ID, Burke B, Freese D, Sharp HL, Kim Y. The pathologic spectrum of the nephropathy associated with alpha 1-antitrypsin deficiency. Human pathology. 1992 Jan;23(1):57-62. PubMed PMID: 1544671.
6. Birrer P, McElvaney NG, Chang-Stroman LM, Crystal RG. Alpha 1-antitrypsin deficiency and liver disease. Journal of inherited metabolic disease. 1991;14(4):512-25. PubMed PMID: 1749216.
7. Childers RE, Ahn J. Diagnosis of Alcoholic Liver Disease: Key Foundations and New Developments. Clinics in liver disease. 2016 Aug;20(3):457-71. PubMed PMID: 27373609.

