Introduction
Cryoglobulins are immunoglobulins that precipitate at low temperatures and redissolve after rewarming18. Brout et al classification, based on the clonality of the immunoglobulins, presents a good correlation between clinical manifestations and associated etiologies5,6. Type I are pure monoclonal immunoglobulins, usually IgG or IgM and are essentially associated with B-cell-proliferative disorders. Type II consists of polyclonal IgG with monoclonal IgM. Type III cryoglobulins are a mixture of polyclonal IgG and IgM1,2,47. Types II and III are generally referred to as mixed cryoglobulinemias and present rheumatoid factor activity. They develop secondarily to infections (most importantly hepatitis C virus HCV), autoimmune or neoplastic disorders16,8. Table 1 presents some of the clinical conditions/agents that may be associated with cryoglobulinemia. When no etiology is identified the cryoglobulinemia is designated as essential2,4,5,7.
Cryoglobulins can cause tissue damage either by hyperviscosity or by vascular inflammation mediated by immune complex depositions and complement fixation1,4,5. Type I cryoglobulinemia patients tend to present clinical manifestations related to complications of hyperviscosity whereas types II and III patients usually display manifestations caused by small-medium vessel vasculitis - Cryoglobulinemia vasculitis (CryoVas)1,47.
It is believed that 2% to 50% of the patients develop symptoms5.Furthermore, clinical expression is extremely variable as CryoVas can involve a variety of tissues (skin, kidneys, joints, lungs, peripheral nerve system or gut) with symptoms that can range from mild (arthralgia) to fulminant life-threatening (widespread vasculitis)1,2,6,8. Meltzer triad purpura, arthralgia and fatigue - is present in 25% to 80%1,5,6. The skin is the most frequently damaged organ (55% to 100% of the patients) with palpable purpura of the lower extremities as the most frequent sign1,2,5,6. Raynauds phenomenon (RF) and acrocyanosis can occur and evolve to digital ulceration / acral ischaemia1,2,5,6,8.
Case Description
A 63-year-old female was referred to an Internal Medicine consultation for possible autoimmune disease. She had a 3-month history of extremally painful digital ulcers, beginning in winter, complicating a RF that started 2 years before. Previous Vascular Surgery consultation had ruled out thromboangiitis obliterans (Buergers disease) after extensive study. There was no clinical improvement with smoking cessation. RF was biphasic and symmetrical to both hands and feet. The patient also referred involuntary weight loss and fatigue. She reported no other symptoms.
The patient had a past medical history of hypertension and depression under treatment with losartan 50 mg q.b., clomipramine 25 mg q.b., and aspirin 100 mg q.b..
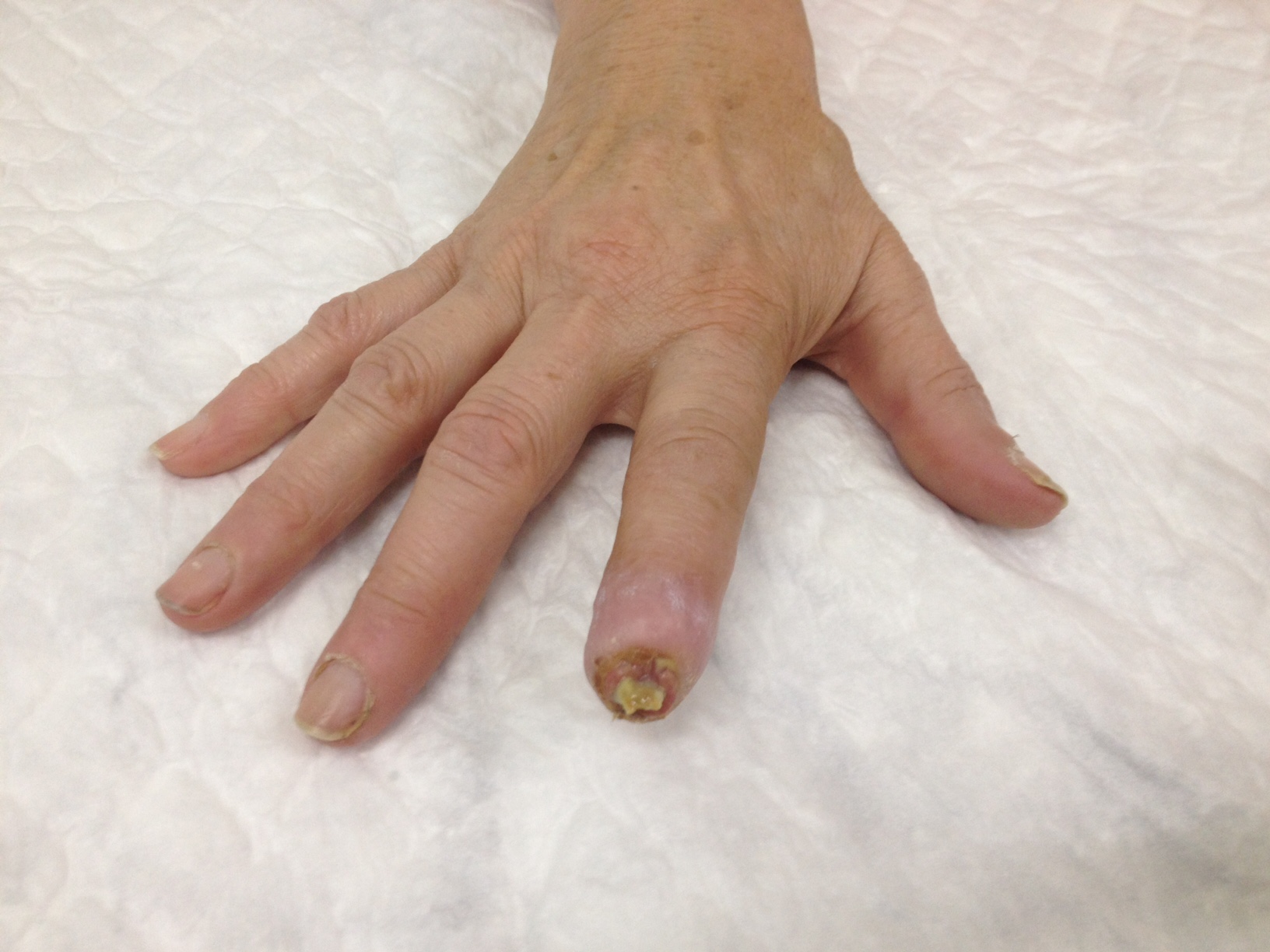
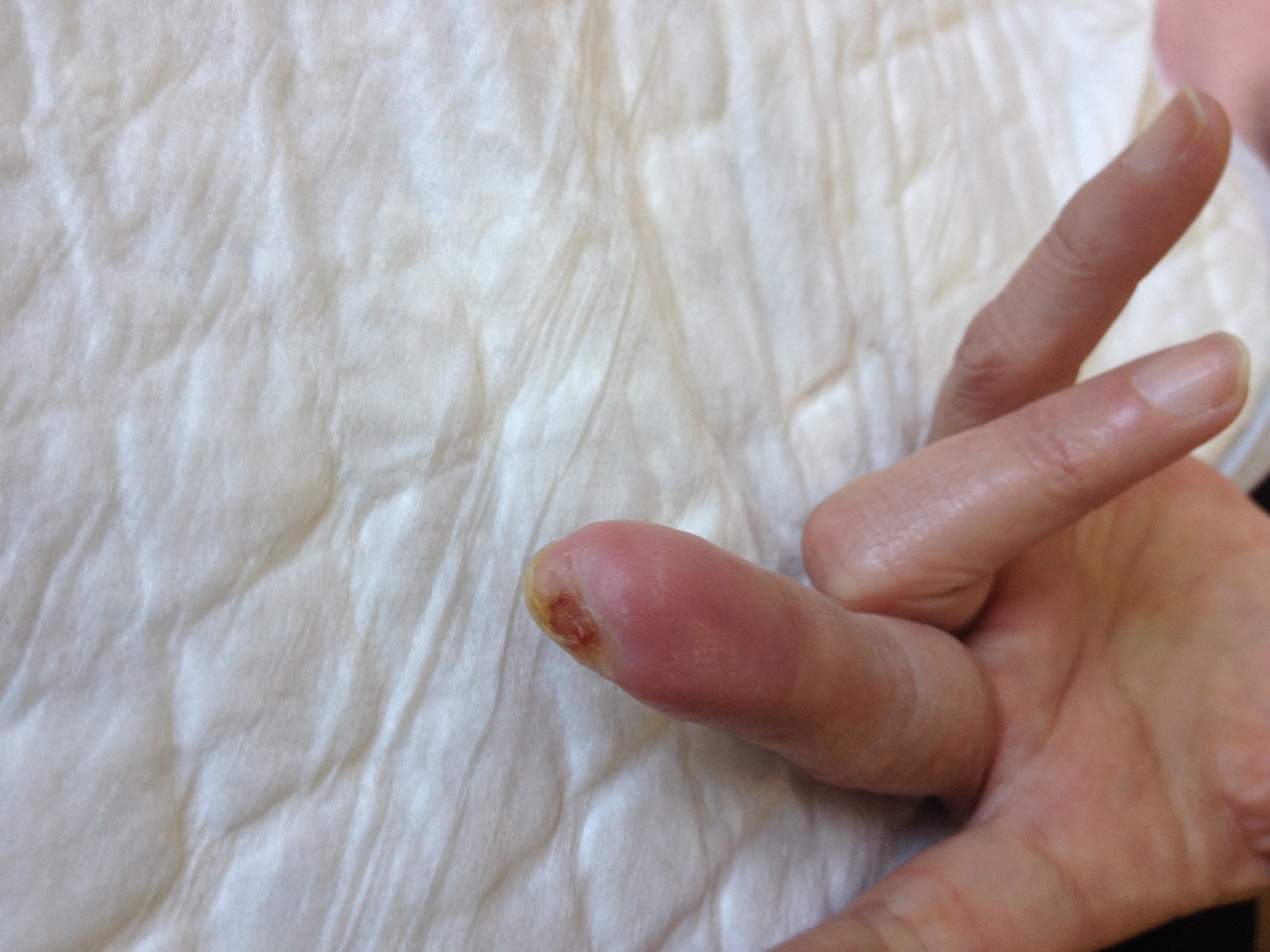
Physical examination revealed severe ulceration of the distal phalanx of the second right finger with bone exposure (figure 1) and small punctate necrosis of the distal phalanx of the third right and left fingers (figure 2 left finger). None of the ulcerations were infected but all were extremely painful. The remaining clinical observation was unremarkable.
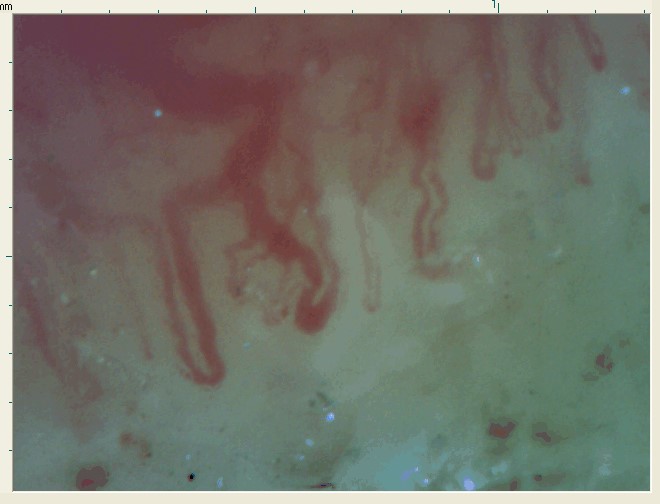
Laboratory results showed a rheumatoid factor of 27 UI/mL (reference range < 20 UI/mL) and the presence of cryoglobulins compatible with cryoglobulinemia type II: 0.9 mg/dL of polyclonal IgG and 4.5 mg/dL of monoclonal IgM. Nailfold capillaroscopy revealed giant capillaries, capillary hemorrhages, and mild disorganization of the capillary architecture (figure 3).
Complete blood cell count, serum biochemistry (including complement C3 and C4), urinalysis and thyroid hormonal studies were normal. Clinical relevant serologies were negative namely HCV, hepatitis B virus (HBV), human immunodeficiency virus 1 and 2 (HIV 1 and 2) and Interferon Gamma Release Assay (IGRA). Serologic tests for autoantibodies were negative including anti-Ro/SSA and anti-La/SSB antibodies (anti-Sjögrens syndrome related antigen A and B), ANA (anti-nuclear antibody), ANCA (anti-neutrophil cytoplasmic antibodies), APS (antiphospholipid syndrome antibodies) and ACPA (anti-citrullinated protein antibodies). No suspicious lesions were identified in computed tomography of the neck, chest, abdomen and pelvis as well as in the PET-scan (positron emission tomography). Bone marrow biopsy revealed no abnormalities.
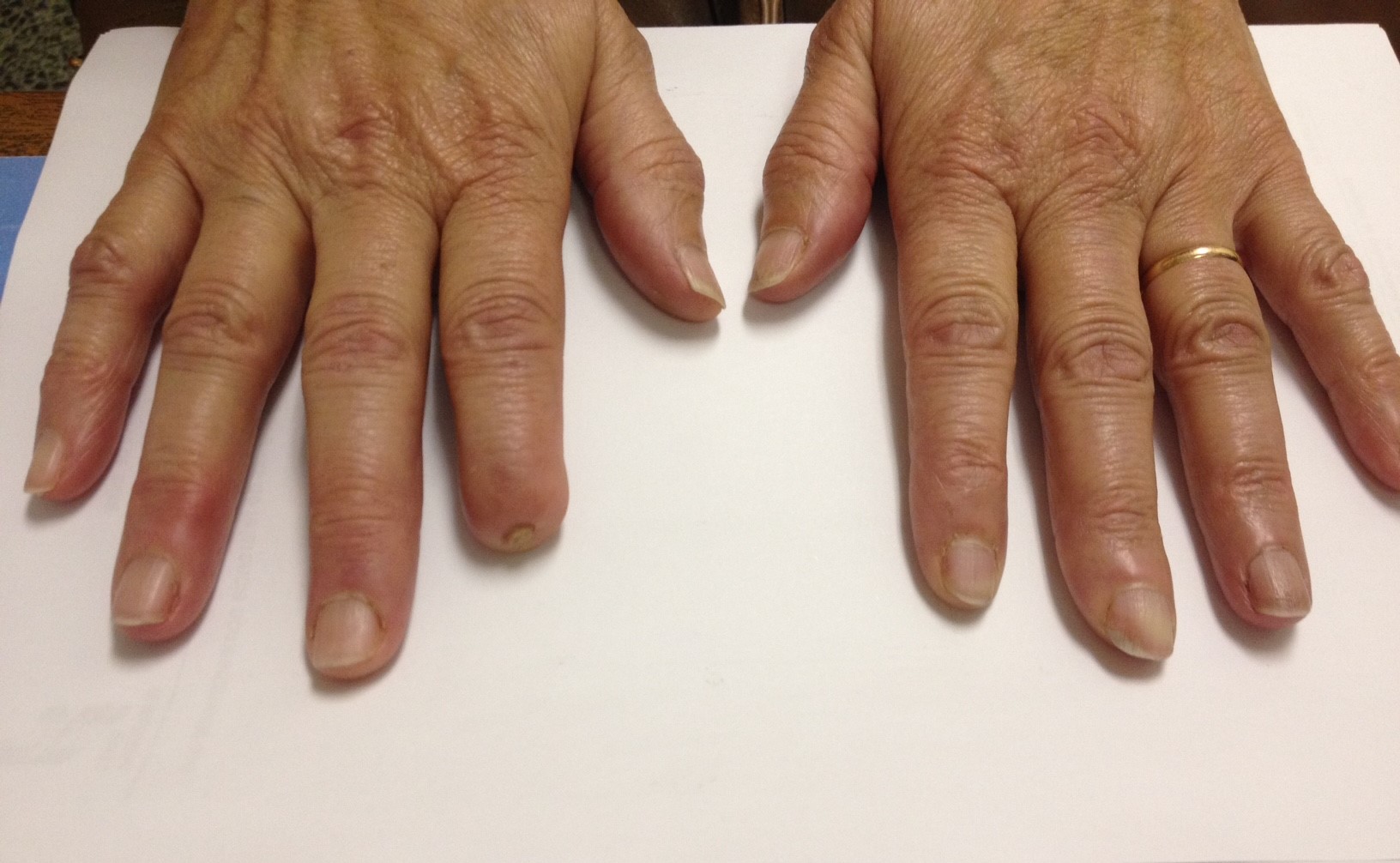
No life-threatening organ damage and no associated etiology was found. Essential type II cryoglobulinemia was assumed and symptomatic treatment was directed to the RF and pain relief. Amlodipine 10 mg, pentoxifylline 400 mg, naproxen 250 mg where started, as well as cold temperature avoidance, use of gloves and skin hydration. After 2 months, the small digital ulcers had healed and the largest one cured after 6 months (figures 4 and 5) with no recurrence after 1 year. Serologies were repeated and remained negative.
Discussion
HCV is the predominant cause of CryoVas, accounting for roughly 80% of the cases14,6,7,9. This identification allowed a better understanding of the disorder1,10. However, as the association was only identified in 199111, most previous studies report results of heterogeneous populations (with or without HCV)2,6,7and most recent studies derive from HCV-positive populations. Thus, data on presentation, therapeutic management and prognosis of non-HCV CryoVas patients is limited3,4. Initiated in 2010, the French CryoVas survey (a national retrospective study) has brought some new insight on non-HCV CryoVas2,3,7.
Essential cryoglobulinemia (EC), as was the case of our patient, accounts for nearly 10% of all patients (up to 25% in non-HCV populations)4,5. Mostly, therapeutic management of CryoVas takes into consideration the underlying disease, the predominant etiopathogenic mechanism (vasculitis vs hyperviscosity) and the severity of the disease1,2,4,9,10. In EC, the absence of etiology and population-orientated studies implies that the best therapeutic management has yet to be defined and treatment usually just involves symptomatic relief2. Mild-to-moderate CryoVas treatment may include resting, cold temperature avoidance, nonsteroidal anti-inflammatory drugs, colchicine and disulone (dapsone and ferrous oxalate)2,4,8. Severe CryoVas can be treated with a combination of corticosteroids, immunosuppressants (rituximab, cyclophosphamide, azathioprine or mycophenolate mofetil) or therapeutic plasma exchange2,4,5,8. Considering that our patient did not present any severe organ damage, conservative treatment directed to RF was our option.
CryoVas presents significant morbidity and mortality10. Prognosis relates to vital organ damage, underlying disease and comorbidities2,4,5,7,8. Generally patients have a worse 10-year survival rate than general population with 15% developing life-threatening complications, although roughly half of the patients never develop vital organ involvement5,8.
CryoVas is already considered a rare disorder despite the absence of adequate epidemiological studies1,5,8. With the development of new therapeutic options for HCV, prevalence of the disorder will eventually decline even further. This case report highlights the importance of awareness of non-HCV CryoVas and the management of these patients.
Acknowledgements
We gratefully acknowledge and thank Dr. Joana Parra for her expertise and skillful technical assistance.
This work was presented in the V National Congress on Autoimunity | XXIII NEDAI Annual Meeting.
Figura I
Digital ulceration
Figura II
Small punctate necrosis
Figura III
Nailfold capillaroscopy
Figura IV
Evolution
Figura V
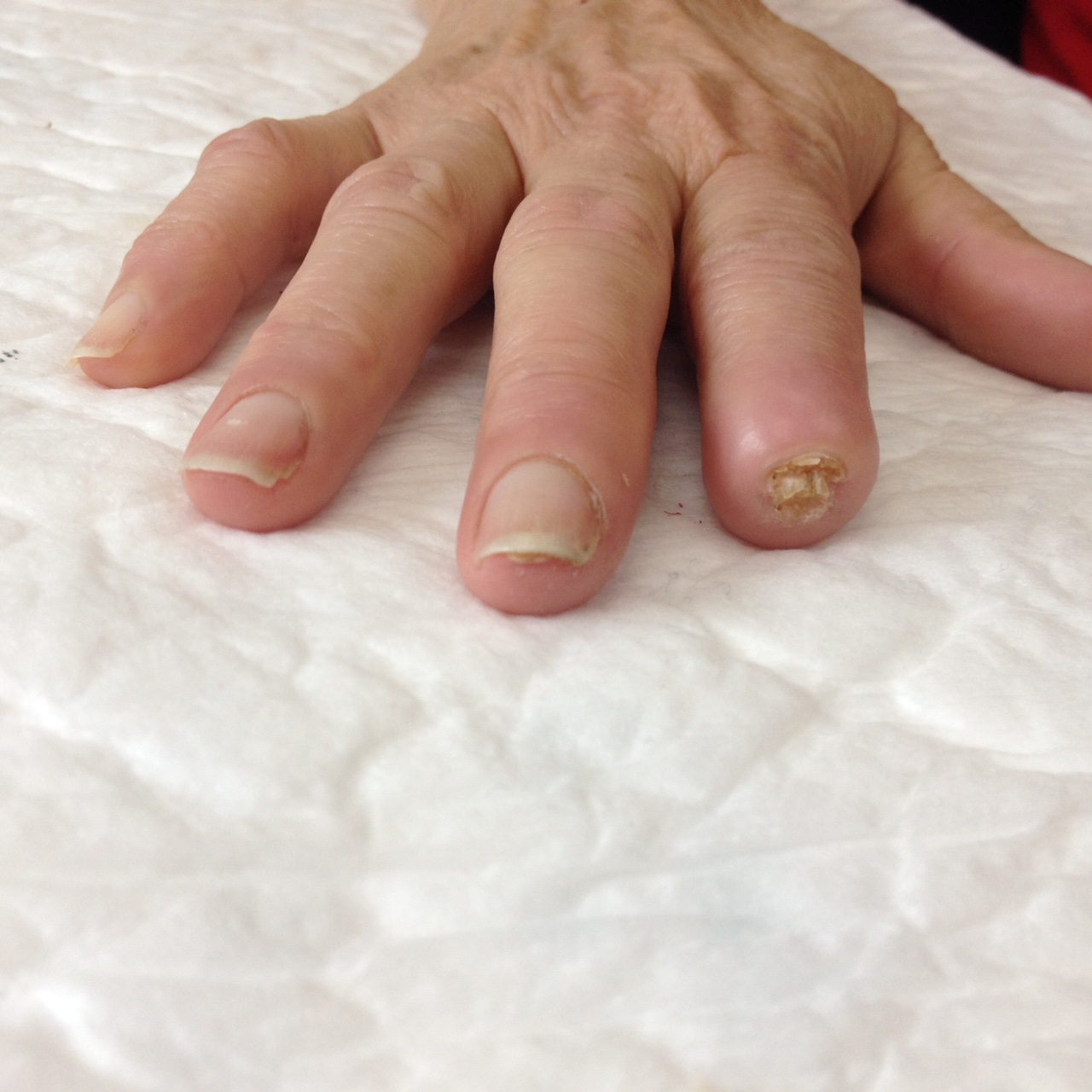
Evolution
Figura VI
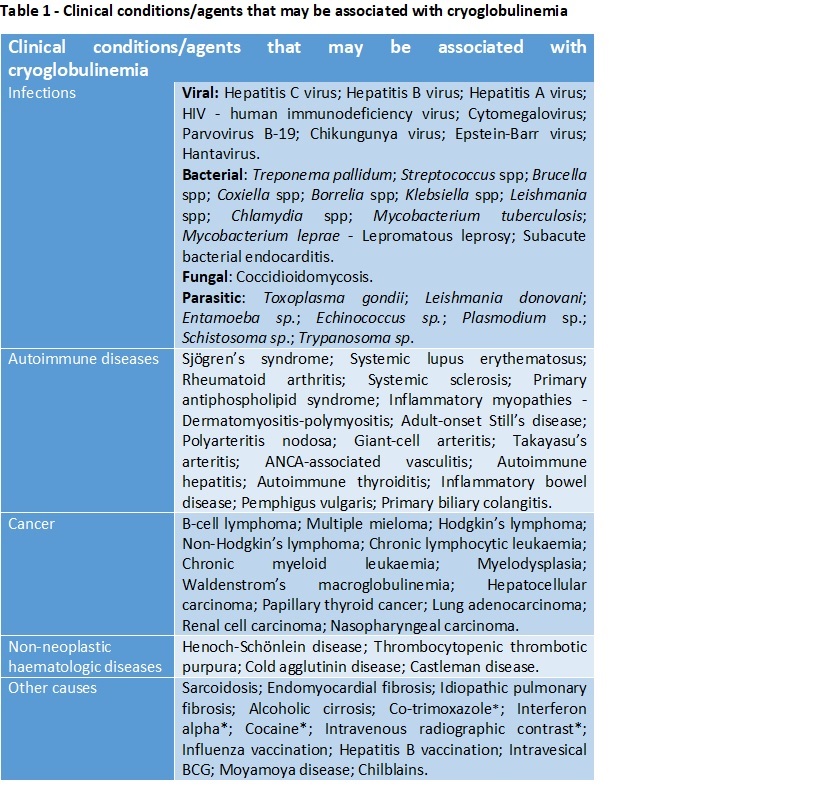
Table 1 List of conditions that may be associated to cryoglobulinemia(5,6); * - Associated with cryoglobulinemic exacerbation.
BIBLIOGRAFIA
References
1. Ghetie D, Mehraban N, Sibley CH. Cold hard facts of cryoglobulinemia. Updates on clinical features and treatment advances. Rheum Dis Clin North Am. 2015;41(1):93108.
2. Terrier B, Cacoub P. Cryoglobulinemia vasculitis: an update. Curr Opin Rheumatol 2013, 25:1018.
3. Terrier B, Marie I, Lacraz A, Belenotti P, Bonnet F, Chiche L, et al. Non HCV-related infectious cryoglobulinemia vasculitis: Results from the French nationwide CryoVas survey and systematic review of the literature. J Autoimmun. 2015;65:7481.
4. Perez-Alamino R, Espinoza LR. Non-infectious cryoglobulinemia vasculitis (CryoVas): Update on clinical and therapeutic approach. Curr Rheumatol Rep. 2014;16(5).
5. Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet. 2012;379(9813):34860.
6. Tedeschi A, Baratè C, Minola E, Morra E. Cryoglobulinemia. Blood Rev. 2007;21(4):183200.
7. Terrier B, Krastinova E, Marie I, Launay D, Lacraz A, Belenotti P, et al. Management of noninfectious mixed cryoglobulinemia vasculitis: Data from 242 cases included in the CryoVas survey. Blood. 2012;119(25):59966004.
8. Takada S, Shimizu T, Hadano Y, Matsumoto K, Kataoka Y, Arima Y, et al. Cryoglobulinemia (Review). Molecular Medicine Reports. 2012; 6:3-8.
9. Elefante E, Monti S, Bond M, Lepri G, Quartuccio L, Talaricor, et al. One year in review 2017: systemic vasculitis. Clinical and Experimental Rheumatology. 2017;35 (Suppl. 103):S5-S26.
10. Cacoub P, Comarmond C, Domont F, Savey L, Saadoun D. Cryoglobulinemia Vasculitis. The American Journal of Medicine. 2015; 128(9):950-955.
11. Ferri C, Greco F, Longombardo G, Palla P, Moretti A, Marzo E, et al. Antibodies to hepatitis C virus in patients with mixed cryoglobulinemia. Arthritis Rheum. 1991;34(12):160610.

