Introdução
O tromboembolismo venoso (TEV), que tem como apresentações clínicas mais frequentes a trombose venosa profunda (TVP) e a embolia pulmonar (EP), constitui uma doença comum e evitável, frequentemente subdiagnosticada, representando uma das três principais causas de morte cardiovascular a nível mundial, a par do enfarte agudo do miocárdio e do acidente vascular cerebral1. Com uma incidência anual, a nível Europeu, de 100-200 casos por cada 100.000 habitantes, os custos de saúde atribuíveis ao TEV excedem os 3 biliões de euros/ano, sendo a EP responsável por cerca de 370.000 mortes anualmente1,2.
A sua fisiopatologia é complexa, envolvendo a parede das veias e a sua lesão por factores extrínsecos (trauma, cirurgia) ou intrínsecos (citoquinas inflamatórias), os componentes celulares do sangue e sua interacção com a parede venosa, a estase venosa e o desequilíbrio entre os sistemas anticoagulante e procoagulante.
As trombofilias, definidas como a tendência para trombose decorrente de alterações hereditárias ou adquiridas da coagulação ou da fibrinólise, conduzem a um estado pró-trombótico.
Na maioria dos casos, as trombofilias hereditárias resultam de mutações de factores da coagulação (Factor V de Leiden, mutação G20210A da protrombina) ou, menos frequentemente, de alterações relacionadas com os inibidores fisiológicos da coagulação (antitrombina, proteína C e proteína S). A elevação marcada da concentração sérica de homocisteína também pode ser responsável por episódios vaso-oclusivos3. A forma genética mais comum de hiperhomocisteinémia resulta da produção de uma variante da metilenotetrahidrofolato reductase (MTHFR) com actividade enzimática reduzida a elevadas temperaturas (termolábil). O gene que codifica esta variante contém uma substituição de citosina por tiamina no nucleótido 677 (677C>T), sendo a forma mutada comum na população geral, com uma frequência estimada entre 5-14%4.
As trombofilias adquiridas são decorrentes de outra situação clínica, como por exemplo síndrome anti-fosfolípido (congénito ou adquirido), neoplasia, imobilização prolongada, cirurgia ortopédica, uso de terapêuticas de reposição hormonal, entre outros.
Caso clínico 1
Homem de 47 anos, electricista. Recorreu ao Serviço de Urgência (SU) por dor tipo moinha na região gemelar direita de início em repouso, com posterior irradiação à face interna da coxa, acompanhado de impotência funcional e de eritema local. Negou traumatismo prévio, dispneia, toracalgia, cansaço fácil ou qualquer outra sintomatologia para além da descrita. Negou hábitos tabágicos ou toxicofílicos. Não apresentava história de cirurgia recente, traumatismo, imobilização prolongada ou viagens de longa duração.
Não apresentava antecedentes patológicos conhecidos nem história familiar de eventos trombóticos.
No exame objectivo realizado na admissão apresentava-se sem sinais de dificuldade respiratória, com pressão arterial de 134/71 mmHg, frequência cardíaca de 78 bpm e temperatura timpânica de 36.5ºC. A auscultação cardiopulmonar e o exame abdominal não revelaram alterações. Foi objectivada dor à palpação na face interna da coxa direita, sem edema periférico, empastamento gemelar ou dor à palpação dos trajectos venosos. O sinal de Homans estava ausente e os pulsos tibiais posteriores e pediosos mantidos e simétricos.
Perante hipótese de TVP foi realizado EcoDoppler venoso dos membros inferiores que confirmou presença de trombo oclusivo da veia safena interna com extensão de 3cm para a veia femoral.
Foi decidido internamento em Enfermaria de Medicina Interna, tendo sido instituída hipocoagulação com enoxaparina com posterior substituição por varfarina. Dada a inexistência de factores precipitantes identificáveis, diversos exames complementares de diagnóstico foram realizados (Tabela 1). Descartada atipia oculta, procedeu-se ao estudo de trombofilias (Tabela 2), em ambulatório, destacando-se: doseamentos normais de antitrombina, proteína S, factor V de Leiden, homocisteína, mutação da protrombina G20210A, anticorpos anti-nucleares (ANA), anticoagulante lúpico, anticorpo anti-cardiolipina e anticorpo anti-β2 glicoproteína. No que respeita à proteína C detectou-se diminuição da sua actividade funcional (41%). A pesquisa das mutações no gene MTHFR, revelou que o doente era portador das variantes 677C>T (genótipo C/T) e 1298A>C (genótipo A/C), ambas em heterozigotia.
O doente mantém seguimento em ambulatório, sem recorrência de evento trombótico, tendo sido tomada decisão de prolongar hipocoagulação até aos 6 meses em função de factores pro-trombóticos identificados.
Caso clínico 2
Mulher de 20 anos, costureira. Obesa, com antecedentes pessoais e familiares irrelevantes, não apresentava hábitos tabágicos ou toxicofílicos, encontrando-se sob anticonceptivo oral (Mercilon®).
Em contexto de dor de instalação súbita em repouso, tipo moinha, na região gemelar esquerda, acompanhada de edema e impotência funcional, recorreu ao SU. Colocada hipótese diagnóstica de tendinite, teve alta medicada sintomaticamente. Reavaliada dias depois em consulta de Medicina do Trabalho, realizou ecografia de partes moles, sem Doppler, que corroborou a hipótese diagnóstica veiculada. Após primeira sessão de programa de Fisioterapia, desenvolveu toracalgia anterior esquerda com irradiação ao dorso e características pleuríticas, recorrendo novamente ao SU. Não apresentava palpitações, lipotímia ou síncope.
No exame objectivo realizado na admissão, apresentava-se polipneica em ar ambiente, com discurso entrecortado, frequência respiratória de 35 cpm e SatO2 de 93%. Encontrava-se normotensa (120/60 mmHg), taquicárdica (130 bpm) e febril (39ºC). A auscultação cardiopulmonar revelou murmúrio vesicular diminuído nas bases, com semiologia abdominal sem alterações. Não apresentava edema periférico, empastamento gemelar, dor à palpação dos trajectos venosos ou sinal de Homans.
Os exames laboratoriais iniciais revelaram elevação de D-dímeros (4.81 mg/L) e insuficiência respiratória parcial (pH 7.45 | pCO2 37 mmHg | pO2 62 mmHg | HCO3- 25.7 mmol/L | SatO2 92%), observando-se apagamento do seio costo-frénico esquerdo na telerradiografia torácica. O ECG apresentava ritmo sinusal, FC 82 ppm, sem padrão S1Q3T3 e sem sinais de sobrecarga cardíaca direita.
Perante suspeita de EP foi realizada Angiotomografia computorizada torácica que confirmou o diagnóstico, documentando trombo ´em sela´ na divisão pulmonar (Fig. 1). Não existindo critérios para trombólise foi iniciada hipocoagulação com enoxaparina, decidindo-se internamento ao cuidado da Medicina Interna.
Integrado no estudo complementar (Tabela 3) foi realizado EcoDoppler venoso dos membros inferiores, que permitiu a identificação de trombo femoro-popliteu esquerdo parcialmente recanalizado.
Após transição para varfarina procedeu-se a alta clínica com indicação para suspensão de anticonceptivo oral.
Pese embora os factores de risco para TEV identificados à admissão (imobilidade condicionada pela actividade profissional, obesidade e toma de anticonceptivo oral), em função da idade jovem e extensão de evento trombótico decidiu-se pela realização de estudo de perfil pró-trombótico (Tabela 2) em ambulatório, de que se destaca: doseamentos normais de antitrombina, proteína C, proteína S, factor V de Leiden, homocisteína, mutação da protrombina G20210A, ANA, anticoagulante lúpico, anticorpo anti-cardiolipina e anticorpo anti-β2 glicoproteína. A pesquisa das mutações no gene MTHFR, revelou doente portadora da variante 677C>T em homozigotia (genótipo T/T).
Também neste caso foi decidido prolongar hipocoagulação até aos 6 meses em função da extensão do evento tromboembólico, trombofilia identificada e evidência ecográfica de recanalização incompleta aos 3 meses. A doente mantém seguimento desde então, sem recorrência de TEV.
Discussão
No primeiro caso clínico, assumiu-se a existência de trombofilia hereditária. Para além da diminuição da actividade funcional da proteína C, o doente era ainda portador das variantes 677C>T (genótipo C/T) e 1298A>C (genótipo A/C) do gene MTHFR, em heterozigotia. Qualquer uma das variantes pesquisadas do gene MTHFR torna a enzima MTHFR termolábil e, consequentemente, com menor actividade. A heterozigotia composta para estas variantes pode associar-se a níveis plasmáticos ligeira ou moderadamente aumentados de homocisteína, particularmente se se verificar défice concomitante de vitamina B6, B12 ou ácido fólico.
No segundo caso, que se assume resultar de uma trombofilia adquirida em função dos factores de risco identificados na anamnese, a investigação complementar documentou a presença de polimorfismo genético potencialmente contribuinte para o risco tromboembólico variante 677C>T (genótipo T/T). Sendo esta variante, particularmente em homozigotia, responsável por um decréscimo de actividade mais evidente, pode também ela estar associada a níveis plasmáticos aumentados de homocisteína, particularmente se acompanhada de défice dos cofactores previamente enumerados.
A hiperhomocisteinémia encontra-se descrita na literatura enquanto factor de risco de TEV4. Entre as causas genéticas de hiperhomocisteinémia encontram-se os polimorfismos do gene MTHFR supradescritos. Contudo, estes polimorfismos apresentam uma prevalência de aproximadamente 11% na população caucasiana, na maioria dos casos (tal como nos que aqui descrevemos) não condicionando alterações nos níveis de homocisteína, pelo que o risco atribuível de TEV é de apenas 1 a 2%5. Não conferindo um risco trombótico significativo na ausência de outros factores trombofílicos, as recomendações actuais não favorecem o seu doseamento.
Existem, contudo, alguns estudos que identificam a S-adenosilhomocisteína enquanto um marcador mais sensível de eventos tromboembólicos6. Sendo um percursor da homocisteína, postula-se que se possa encontrar elevado mesmo na presença de níveis plasmáticos normais de homocisteína. Todavia, mais estudos serão necessários para esclarecer a pertinência da pesquisa de mutações no gene MTHFR na presença de doseamentos de homocisteína normais.
Do ponto de vista estritamente teórico, a identificação de uma trombofilia hereditária possibilitaria a instituição de hipocoagulação profilática. Se por um lado não existe evidência científica que justifique os riscos desta abordagem nos indivíduos assintomáticos (isto é, que nunca sofreram um TEV), por outro, esta opção poderá ser considerada em algumas situações particulares de potencial alto-risco, como por exemplo cirurgia major, trauma, imobilização prolongada, gravidez ou puerpério7.
A evidência científica actual recomenda pelo menos 3 meses de hipocoagulação oral após um primeiro episódio de TEV. Todavia, a duração ideal da terapêutica hipocoagulante (de 3 meses a ad æternum) permanece envolta em intenso debate8-9, pelo que deverá ser equacionada de forma individualizada.
Quadro I
Tabela 1
| Caso Clínico 1 |
| MCDTs |
| EcoDoppler venoso dos membros inferiores |
| TC Torácica |
| TC Abdominal |
| TC Pélvica |
| Ecografia prostática transrectal |
| Estudo de perfil pró-trombótico |
Exames complementares de diagnóstico realizados, Caso Clínico 1
Quadro II
Tabela 2
| Casos Clínicos 1 e 2 |
| Estudo pró-trombótico |
| Antitrombina |
| Proteína C |
| Proteína S |
| Factor V de Leiden |
| Homocisteína |
| Mutação da protrombina G20210A |
| Mutação do MTHFR |
| Anticoagulante lúpico |
| Anticorpo anti-cardiolipina (IgM e IgG) |
| Anticorpo anti-β2 glicoproteína |
Estudo pró-trombótico, Casos Clínicos 1 e 2
Quadro III
Tabela 3
| Caso Clínico 2 |
| Exames complementares de diagnóstico realizados |
| EcoDoppler venoso dos membros inferiores |
| AngioTC torácica |
| Ecocardiograma |
| Estudo de perfil pró-trombótico |
Exames complementares de diagnóstico realizados, Caso Clínico 2
Figura I
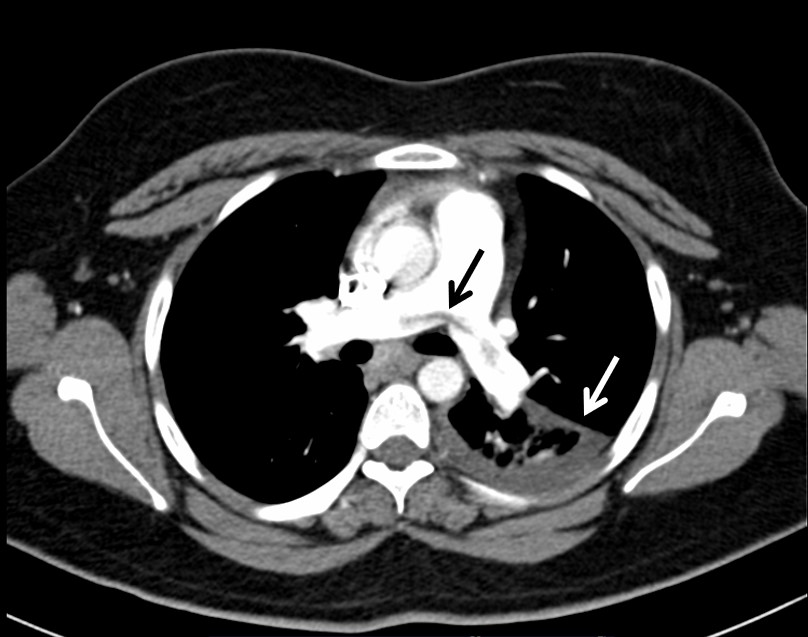
AngioTC. Trombo em sela na divisão pulmonar (seta preta). Derrame pleural esquerdo e consolidação da pirâmide basal adjacente (seta branca).
Figura II
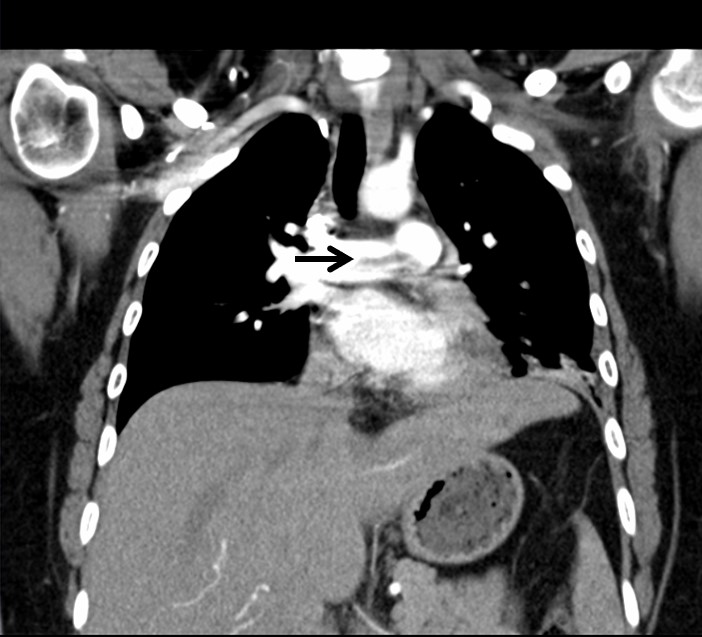
AngioTC. Trombo em sela na divisão pulmonar (seta preta).
BIBLIOGRAFIA
1 Goldhaber SZ. Deep Venous Thrombosis and Pulmonary Thromboembolism. In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J. Harrison´s Principles of Internal Medicine, 18th Edition. New York: McGraw-Hill, 2012: 4414-4433.
2 D´Amico EA. Trombofilia: quando suspeitar e como investigar? Rev. Assoc. Med. Bras. Vol.49 nº.1 São Paulo Jan./Mar. 2003.
3 Rosenson RS, Kang DS. Overview of homocysteine. UpToDate 2014.
4 Bauer KA. Screening for inherited thrombophilia in asymptomatic individuals. UpToDate 2014.
5 Zhu T, et al. Venous thromboembolism: risk factors for recurrence. Arterioscler Thromb Vasc Biol. 2009; 29: 298-310.
6 Donadini MP, Ageno W. Which patients with unprovoked VTE should receive extended anticoagulation? the minority. Journal of Thrombosis and Thrombolysis, April 2011, Volume 31, Issue 3, pp 301-305.

