

Introdução:
O Angioedema hereditário (AEH) é uma doença genética rara, autossómica dominante, categorizada como uma imunodeficiência primária. Caracteriza-se clinicamente por episódios recorrentes, localizados e autolimitados de angioedema (edema das camadas subcutânea ou submucosa), com envolvimento preferencial da face, extremidades, genitais, trato respiratório superior ou gastrointestinal.1-3 O edema que caracteriza a doença é independente da gravidade, assimétrico e godet negativo.4
Episódios de angioedema intestinal ocorrem em mais de 85% dos casos. Podem ser a única manifestação da doença e apresentar-se com dor abdominal súbita e intensa, frequentemente mal diagnosticada e abordada como um quadro de abdómen agudo.2,5,6
Apresentamos um caso de AEH diagnosticado aos 62 anos, após múltiplos internamentos por gastroenterites recorrentes.
Caso clínico:
Homem de 62 anos, caucasiano, com antecedentes de hipertensão arterial, medicado com lisinopril há vários anos.
Apresenta-se por quadro com 4 horas de evolução de diarreia (7 dejeções aquosas, sem sangue ou muco), vómitos biliares e dor abdominal difusa, intensa, tipo cólica. Sem febre, sintomas constitucionais, respiratórios ou alterações cutâneas. Sem contexto epidemiológico de risco. Admitido na sala de emergência por choque hipovolémico, com disfunção multiorgânica.
Ao exame objetivo, apirético, sem evidência de irritação peritoneal, edema subcutâneo, urticária ou sinais de dificuldade respiratória. Internado no Serviço de Medicina Intensiva com diagnóstico presuntivo de gastroenterite aguda, sob fluidoterapia e aminas, com recuperação completa do quadro clínico em 48h.
Nos quatro meses seguintes apresentou seis recorrências da mesma clínica, com necessidade de internamento em quatro dos episódios, sempre com rápida recuperação, apenas sob tratamento de suporte com fluidoterapia. Apresentou perda ponderal de 8% nesse período e desenvolveu quadro depressivo.
O estudo exaustivo dirigido às possíveis etiologias de diarreia crónica, excluiu síndrome de má absorção, doença infeciosa, metabólica/endócrina, autoimune (DAI) e neoplásica. A Tomografia computorizada revelou ascite de reduzido volume em fase aguda de doença (confirmada resolução em exame de reavaliação), sem espessamento de ansas intestinais. Realizada colonoscopia sem alterações macroscópicas e histologia (13 biópsias do íleon ao recto) com acúmulo focal de linfócitos e alguns eosinófilos no córion, mas sem critérios para colite microscópica (linfocítica) ou gastroenterite eosinofílica.
De todo o estudo mantinha-se por explicar um resultado inicial: diminuição do fator C4 do complemento (5 mg/dL; N: 15-53). Foi então valorizado, avançando-se a hipótese diagnóstica de Angioedema isolado.
Revista a anamnese e só quando questionado direta e veemente, afirmou pela primeira vez episódios recorrentes e autolimitados de edema das mãos e pés, desencadeados por traumatismos, desde há mais de 20 anos, compatíveis com angioedema subcutâneo. Confirmou ainda, história familiar de angioedema hereditário tipo 1 (filho e neto).
Realizados testes quantitativo e qualitativo do inibidor de C1 (C1INH), ambos diminuídos (7,2 mg/dL - N: 21-39, e 12% - N 80-125%, respetivamente) em duas avaliações separadas por dois meses. Confirmado, assim, o diagnóstico de AEH tipo 1.
Suspendeu-se o lisinopril habitual, sem expectável resolução do angioedema, mas com diminuição do número de episódios. Excluídas outras causas de angioedema (outros fármacos e história de alergia alimentar ou ambiental corroborada por IgE total e triptase normais). Dado o início tardio dos sintomas, excluídas também doenças associadas a angioedema adquirido com défice de C1INH (AEA-C1INH) e realizado teste quantitativo de C1q que foi normal.
Iniciou seguimento por imunoalergologia em centro de referência de tratamento de AEH e terapêutica com androgénio atenuado (Danazol 100 mg/dia). Mantém-se atualmente assintomático, sem reações adversas identificadas após 3 anos de tratamento.
Discussão:
O AEH, com uma prevalência estimada de 1:50.000,2,5 é responsável por uma minoria de todos os casos de angioedema, manifestação clínica com múltiplas outras etiologias descritas (Fig. 1).7,8
Atualmente classifica-se em quatro tipos: AEH tipo 1 (85% dos casos) e tipo 2 resultam de défice quantitativo ou qualitativo/funcional de C1INH, respetivamente, e em conjunto classificam-se por AEH com défice de C1INH (AEH-C1INH); os outros 2 tipos caracterizam-se por estudos do complemento normais e dividem-se em AEH associado a mutação do fator XII de coagulação (AEH-FXII) e AEH de causa desconhecida (AEH-U).3,9 Em 2017 foi identificada uma possível mutação no gene da angiopoietina-1, que pode representar um quinto tipo de AEH.10 AEH-FXII e AEH-U são clinicamente semelhantes ao AEH-C1INH3,8 e a sua apresentação detalhada está fora do âmbito deste artigo.
Os tipos 1 e 2 são clinicamente indistinguíveis e caracterizam-se por episódios recorrentes de angioedema já descritos, tipicamente não associados a urticária ou prurido, como no caso clínico apresentado.4,9 Habitualmente os sintomas começam na infância ou adolescência, sendo que apenas 5% dos doentes permanecem assintomáticos após os 20 anos.7 Apesar dos avanços recentes no entendimento da doença, está descrito um atraso no diagnóstico de 13-21 anos.4,5,11
A gravidade e frequência dos sintomas é muito variável e imprevisível no próprio doente ao longo do tempo e entre doentes da mesma família, e não se correlaciona com os níveis de C1INH no plasma.1,9 Pelas suas características fisiopatológicas é autolimitado, com resolução habitual em 2-5 dias. No entanto, na ausência de terapêutica adequada, pode ser potencialmente fatal por asfixia secundária a envolvimento laríngeo, pelo que o diagnóstico de AEH é de extrema importância.2,5
As crises abdominais, sintoma isolado em 50% dos casos, podem apresentar-se como desconforto leve ou integrar-se em quadros mais graves e debilitantes como o descrito, com dor tipo cólica, intensa, acompanhada de vómitos e/ou diarreia.6 Devido à semelhança clínica com emergências cirúrgicas, cerca de 30% dos doentes com AEH não diagnosticado acabam por ser submetidos, desnecessariamente, a cirurgia abdominal.7 Salienta-se que leucocitose e sinais de irritação peritoneal podem estar presentes no AEH.6,7 O edema intestinal com a passagem de líquidos para o interstício e cavidade peritoneal pode ser causa de hipovolemia com potencial evolução para choque.4 Tanto o edema da parede intestinal como a ascite são detetados nos exames de imagem durante as crises, com resolução posterior, como descrito neste caso clínico.7,12
Nos episódios anteriores de angioedema relatados pelo doente, havia história de um traumatismo minor prévio, um dos desencadeantes de crises mais frequentemente relatado na literatura.1 Outros fatores precipitantes são: procedimentos estomatológicos ou cirúrgicos, stress, infeções (p.e. Helicobacter pylori7) e fármacos como inibidores da enzima de conversão de angiotensina e anticoncecionais orais, que podem exacerbar a frequência e severidade dos episódios, devendo ser evitados.2,4 Na maioria dos casos não se identifica fator desencadeante, tal como nas crises abdominais mais recentes do doente.1,4
O doseamento sérico de C4 é o exame de rastreio recomendado, por ser sempre baixo. Apenas 2% dos doentes apresentam valores normais nos períodos assintomáticos.4,6
O diagnóstico de AEH deve ser equacionado em doentes com história de dor abdominal e/ou angioedema recorrente, sem urticária, com duração de 2-5 dias sem tratamento, associado a história familiar e com diminuição de C4 sérico. A ausência de história familiar não excluiu o diagnóstico, dado que até 25% dos casos representam mutações de novo.3,9 Na presença de um quadro típico, o défice quantitativo e/ou qualitativo de C1INH em duas avaliações separadas por 1-3 meses, confirma o diagnóstico de AEH-C1INH.3
No caso clínico apresentado salienta-se que o doente nunca valorizou as queixas de angioedema subcutâneo recorrentes, nem a história familiar, só o referindo quando questionado diretamente e de forma dirigida à investigação de um possível AEH. Uma avaliação mais cuidada da história, aliado aos resultados analíticos característicos (Fig. 2) levaram ao diagnóstico.
Dado o início de queixas tardio na idade adulta (acima dos 40 anos) e apesar da história familiar, tornou-se premente excluir AEA-C1INH, assim como doenças linfoproliferativas, Gamapatia Monoclonal e DAI que lhe estão habitualmente associadas.3,9
Pela sua maior prevalência, é sempre necessário excluir a possibilidade de angioedema de causa alérgica.9
No que diz respeito ao tratamento, dado que o mediador responsável pelo angioedema nestes doentes é a bradicinina, a utilização de anti-histamínicos, corticosteróides ou adrenalina é ineficaz.2,4,8
O tratamento agudo de episódios severos passa por concentrado de C1INH purificado ou recombinante, antagonistas do recetor da bradicinina (Icatibant) ou inibidor de calicreína (ecallantide; não disponível em Portugal); em 2ª linha, plasma fresco congelado (PFC).2-4,8,13 Podem ser associados antiespasmódicos para controlo sintomático nas crises abdominais.4
A decisão de instituir profilaxia de longo-prazo deve ser individualizada e de acordo com o impacto na qualidade de vida, o qual depende da frequência, gravidade e localização dos episódios.4 Utilizam-se androgénios atenuados (danazol, estanazol) e em 2ª linha, antifibrinolíticos (ácido aminocapróico, ácido tranexâmico) ou concentrado de C1INH.3,11
Todos os doentes devem realizar profilaxia de curto-prazo com androgénios, concentrado de C1INH ou PFC, antes de situações previsíveis de stress, intervenções estomatológicas ou cirúrgicas.2-4
Está descrita uma taxa de mortalidade de 13% por asfixia7,14, a qual é 3-9 vezes superior nos doentes sem diagnóstico estabelecido, sendo potencialmente evitável se reconhecida a etiologia e tratada adequada e atempadamente.8,14
Conclusão:
As queixas sugestivas de abdómen agudo recorrentes e sem causa identificada, devem alertar para a possibilidade do diagnóstico de AEH, independentemente da idade e da escassez de manifestações cutâneas de angioedema. Na ausência de suspeição de AEH incorre-se no risco de realizar tratamentos desnecessários e ineficazes, com consequente aumento da morbimortalidade e dos custos para a instituição.5,15
Este caso ilustra a importância de estabelecer o diagnóstico etiológico de forma a evitar a recorrência da doença, permitindo também estabelecer o plano terapêutico mais adequado e rastrear precocemente a família.2,5
Figura I
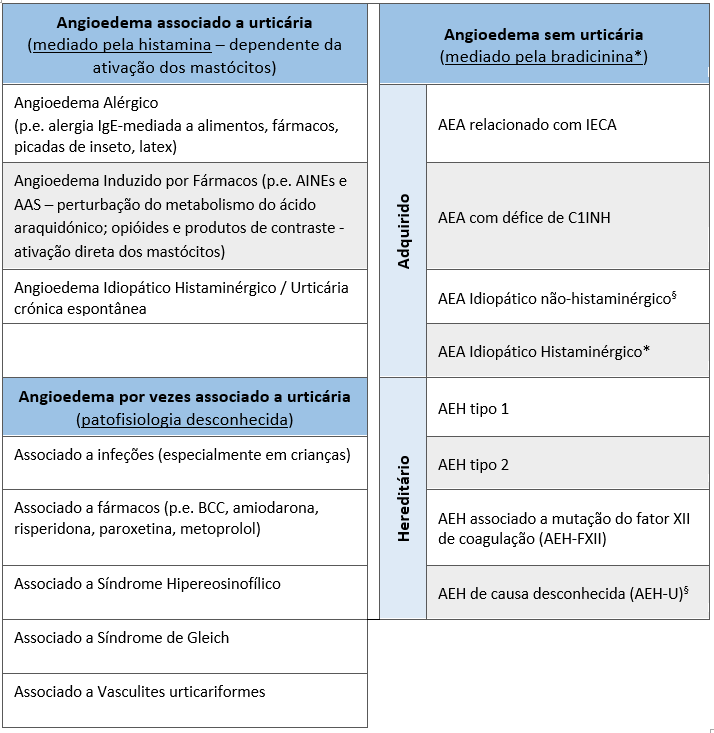
Classificação de Angioedema. *Exceto no AEA Idiopático Histaminérgico em que se presume ser mediado pela histamina dada a resposta aos anti-histamínicos; §Presume-se mediação pela bradicinina, mas ainda não está comprovado. AINEs Anti-inflamatórios não esteroides; AAS Ácido Acetilsalicílico; BCC Bloqueador dos canais de cálcio; AEA Angioedema adquirido; IECA inibidor da enzima de conversão de angiotensina; AEH Angioedema Hereditário; C1INH C1 inibidor.
Figura II
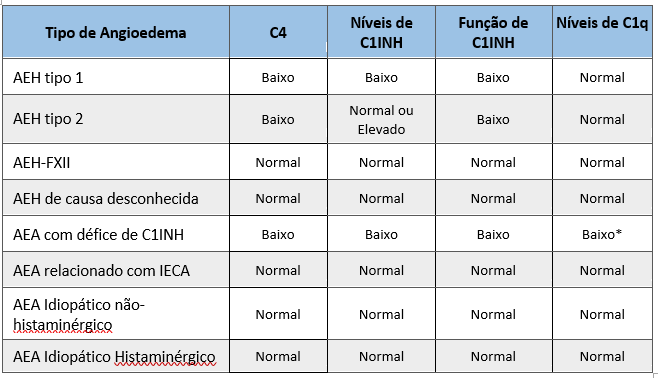
Perfil analítico característico das diferentes formas de Angioedema sem urticária. * C1q encontra-se baixo em >70% dos casos 3; AEH Angioedema Hereditário; AEH-FXII associado a mutação do fator XII de coagulação; C1INH C1 inibidor; AEA Angioedema adquirido; IECA inibidor da enzima de conversão de angiotensina.
BIBLIOGRAFIA
1. Zuraw BL, Christiansen SC. HAE Pathophysiology and Underlying Mechanisms. Clin Rev Allergy Immunol. 2016 Oct;51(2):216-29.
2. Grumach AS, Ferraroni N, Olivares MM, et al. An ABC of the Warning Signs of Hereditary Angioedema. Int Arch Allergy Immunol. 2017;174(1):1-6.
3. Cicardi M, Aberer W, Banerji A, et al on behalf of HAWK, under the patronage of EAACI (European Academy of Allergy and Clinical Immunology). Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy 2014; 69: 602616.
4. Katelaris C, Smith W, Wong M, Jordan A. Australasian Society of Clinical Immunology and Allergy (ASCIA): Position paper on Hereditary angioedema; 2017 [consultado 2017 Set 30]. Disponível em: https://www.allergy.org.au/health-professionals/papers/hereditary-angioedema
5. Zanichelli A, Magerl M, Longhurst H, et al. Hereditary angioedema with C1 inhibitor deficiency: delay in diagnosis in Europe. Allergy Asthma Clin Immunol. 2013 Aug 12;9(1):29.
6. Rubinstein E, Stolz LE, Sheffer AL, et al. Abdominal attacks and treatment in hereditary angioedema with C1-inhibitor deficiency. BMC Gastroenterol. 2014 Apr 9;14:71.
7. Agostoni A, Aygören-Pürsün E, Binkley KE, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol 2004; 114:S51.
8. Bernstein JA, Cremonesi P, Hoffmann TK, Hollingsworth J. Angioedema in the emergency department: a practical guide to differential diagnosis and management. Int J Emerg Med. 2017 Dec;10(1):15.
9. Wu MA, Perego F, Zanichelli A, Cicardi M. Angioedema Phenotypes: Disease Expression and Classification. Clin Rev Allergy Immunol. 2016 Oct;51(2):162-9.
10. Bafunno V, Firinu D, D´Apolito M, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. 2017 Jun 8. pii: S0091-6749(17)30921-1.
11. Gómez-Traseira C, Pérez-Fernández E, López-Serrano MC, et al. Clinical Pattern and Acute and Long-term Management of Hereditary Angioedema Due to C1-Esterase Inhibitor Deficiency. J Investig Allergol Clin Immunol. 2015;25(5):358-64.
12. Gábos G, Dobru D, Mihály E, et al. Recurrent ascites: a need to evaluate for hereditary angio-oedema. Lancet. 2017 Nov 4;390(10107):2119-2120
13. Longhurst HJ, Tarzi MD, Ashworth F, et al. C1 inhibitor deficiency: 2014 United Kingdom consensus document. Clin Exp Immunol. 2015 Jun;180(3):475-83.
14. Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012 Sep;130(3):692-7.
15. Otani IM, Christiansen SC, Busse P, et al. Emergency Department Management of Hereditary Angioedema Attacks: Patient Perspectives. J Allergy Clin Immunol Pract. 2017 Jan - Feb;5(1):128-134.e4.