LEARNING POINTS:
· SS and SLE are characterized by a great heterogeneity in clinical presentation. The coexistence of COP and Evans syndrome with severe thrombocytopenia is rare;
· 2016-ACR/EULAR classification criteria for SS recognize its systemic nature and allowing diagnosis in patients without salivary or ocular glandular symptoms. In some cases, this may result in early identification of SS, providing patients with the opportunity of early treatment
CASE DESCRIPTION
A 25-year-old Caucasian woman was admitted to an Internal Medicine Department due to a history of myalgia, polyarthralgia, dry cough, pleuritic chest pain, vespertine high fever, fatigue and anorexia for 1-month. Polyarthralgia affecting small and large joints (shoulders, metacarpophalangeal, interphalangeal and ankles), was additive, symmetrical, and with typical inflammatory rhythm. Xerophthalmia, xerostomia and weight loss were denied. Her clinical symptoms did not improve after treatment with levofloxacin and ibuprofen.
There was no long-term medication history, recent travel or exposure to occupational and agricultural diseases. Recurrent infections or malignancy were not reported. Family history was negative for autoimmune or neoplastic diseases; however her mother and grandmother had an underinvestigated history of polyarthralgias.
Physical examination revealed petechial rash, spontaneous cutaneous hematomas and bi-basal lung rales. Her molar teeth had extensive caries lesions. The remainder physical examination was normal.
METHODS AND PROCEDURES
Laboratorial data revealed severe thrombocytopenia (4x109/L), microcytic anemia (hemoglobin 10.1g/dl and mean corpuscular volume of 75.7fl and leukopenia (3.4x109/L). Anti-platelet and indirect antiglobulin antibodies were detected. Erythrocyte sedimentation rate was elevated (42mm/h), C-reactive protein (CRP) level of 5mg/L and LDH of 772mg/dL. Liver and kidney function, thyroid hormones, electrolytes and urinary sediment were normal. She tested positive for adenovirus infection by adenovirus antigen test, but was negative for other viral antigen tests. Hematologic malignancies and sarcoidosis were excluded. Immunological study showed mild hypergammaglobulinemia. C3 and C4 were within normal range. Cryoglobulins and rheumatoid factor were not detected. Her autoantibody profile showed positive antinuclear antibodies (titer 1/640, homogeneous fluorescence pattern) with anti-SSA/Ro positivity. Anticyclic citrullinated peptide, double-stranded DNA and Anti-neutrophil cytoplasmic antibodies were negative. Chest radiography and computed tomography (CT) showed bilateral parenchymal infiltrate in pulmonary bases (figure 1 and 2). Schirmer´s test was negative. Salivary gland scintigraphy revealed functional absence of the submandibular glands (figure 3) and salivary gland biopsy revealed focal lymphocytic aggregates, with a focus score of 1 focus/4mm2 (figure 4).
An overlap syndrome was diagnosed, with features of SLE and SS. The patient was treated with prednisone 1mg/Kg daily and hydroxychloroquine. She showed marked clinical improvement and was discharged home on a tapering dose of prednisone.
DISCUSSION
SS and SLE involve multiple organ systems and follow a variable course of illness. The 2016-ACR/EULAR classification criteria for SS was fulfilled in our patient as she exhibited the presence of reduced salivary gland uptake in salivary scintigraphy, focal lymphocytic sialadenitis and a positive serological test for Ro (SSA). Furthermore, the patient also fulfilled the 2012-SLICC classification criteria for SLE as we observed arthritis, haemolytic anaemia, leukopenia, thrombocytopenia and anti-Ro/SSA (4 clinical and 1 immunological criteria). Accordingly, the authors assumed a Sjögren-Lupus Overlap Syndrome.
Anti-SSA/Ro and Anti-SSB/La are the hallmark antibodies in SS, and are present in 4080% of patients, but anti-SSA/Ro is the most specific.1 Patients with anti-SSA/Ro antibodies have the highest prevalence of most systemic, hematologic and immunologic alterations.1 In SLE, anti-Ro/SSA antibodiesmight be present in 30-40% of patients, while anti-dsDNA might be detected in 70-80% of patients.4
Complement activation plays a key role in the pathophysiology of SLE and it is recommended to monitor serum levels of C3 and C4 to assess for disease activity. However, it is important to note that decreased serum complement is not consistently associated with disease flares, as observed in our patient.5
Recently, Yang and co-authors6 reported that SS/SLE patients had an earlier age of onset, higher incidences of arthritis, leukopenia, proteinuria and and lower incidences of xerostomia and interstitial lung disease compared with primary SS patients. These results suggest that patients presenting with this clinical and laboratory characteristics are more likely to develop SS/SLE.
Although systemic manifestations are common for SS and SLE patients, our case describes uncommon pulmonary and hematological manifestations:
- Cryptogenic Organized Pneumoniae and lymphocytic interstitial pneumonia have been reported as initial SS manifestation;1 In SLE it was reported as clinically significant in only 3% and pleural disease in 17% of patients at the time of diagnosis, increasing to 17% and 36%, respectively, over the course of disease.2
- Severe autoimmune thrombocytopenia and haemolytic anaemia, also knows as Evans Syndrome (ES), are haematological complications rarely described (<1% for SS and 2.7% for SLE), suggesting an autoimmune and severe multisystemic disease.1,3
The occurrence of a true overlap between SS and SLE is relatively rare and the ACR/EULAR classification criteria may allow a higher sensitivity to detect such condition, namely in patients without sicca symptoms, the hallmark of SS.
Figura I
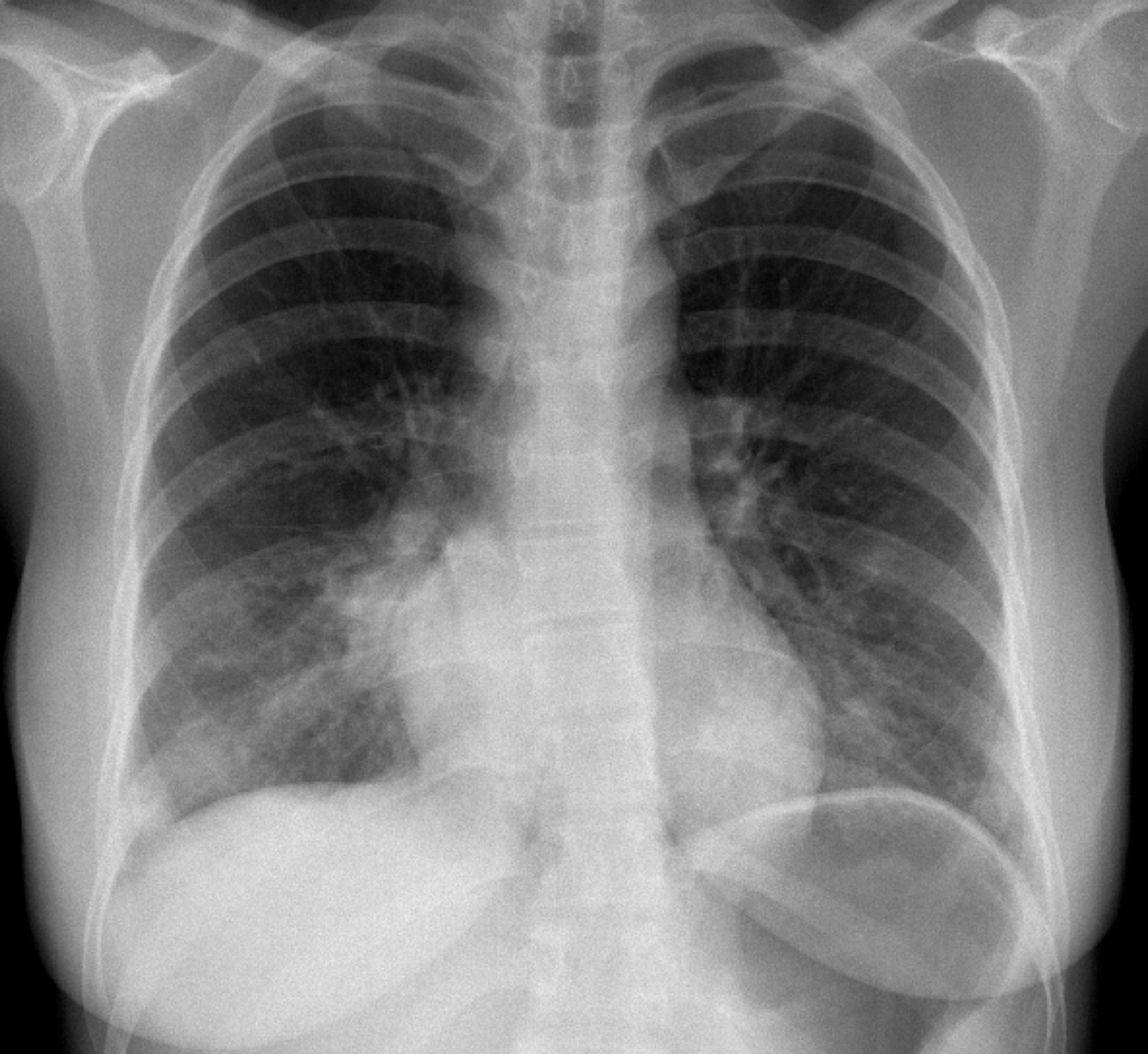
PA chest radiograph with bilateral infiltrates
Figura II
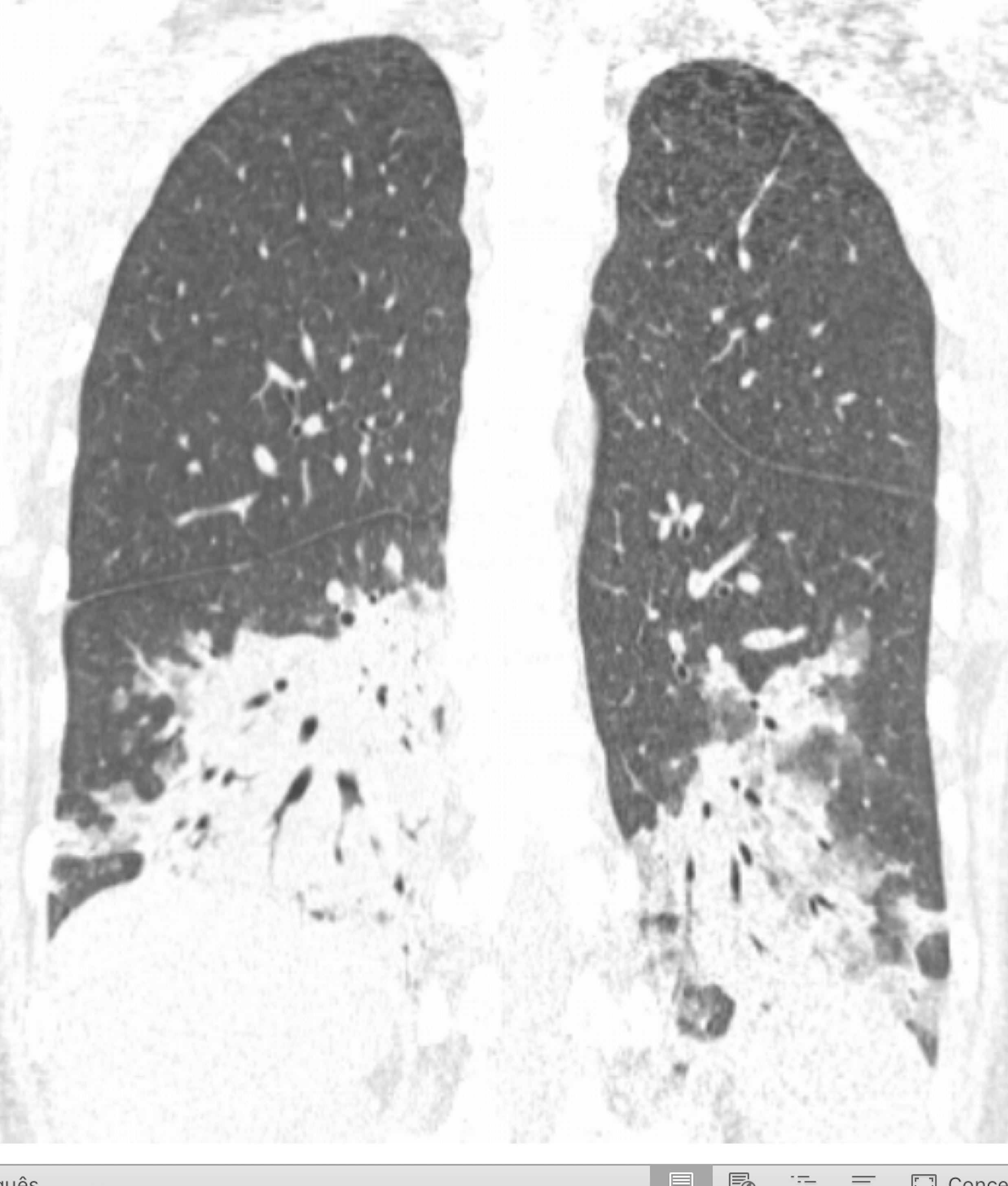
Coronal chest CT scan showing bilateral parenchymal infiltrate in pulmonary bases
Figura III
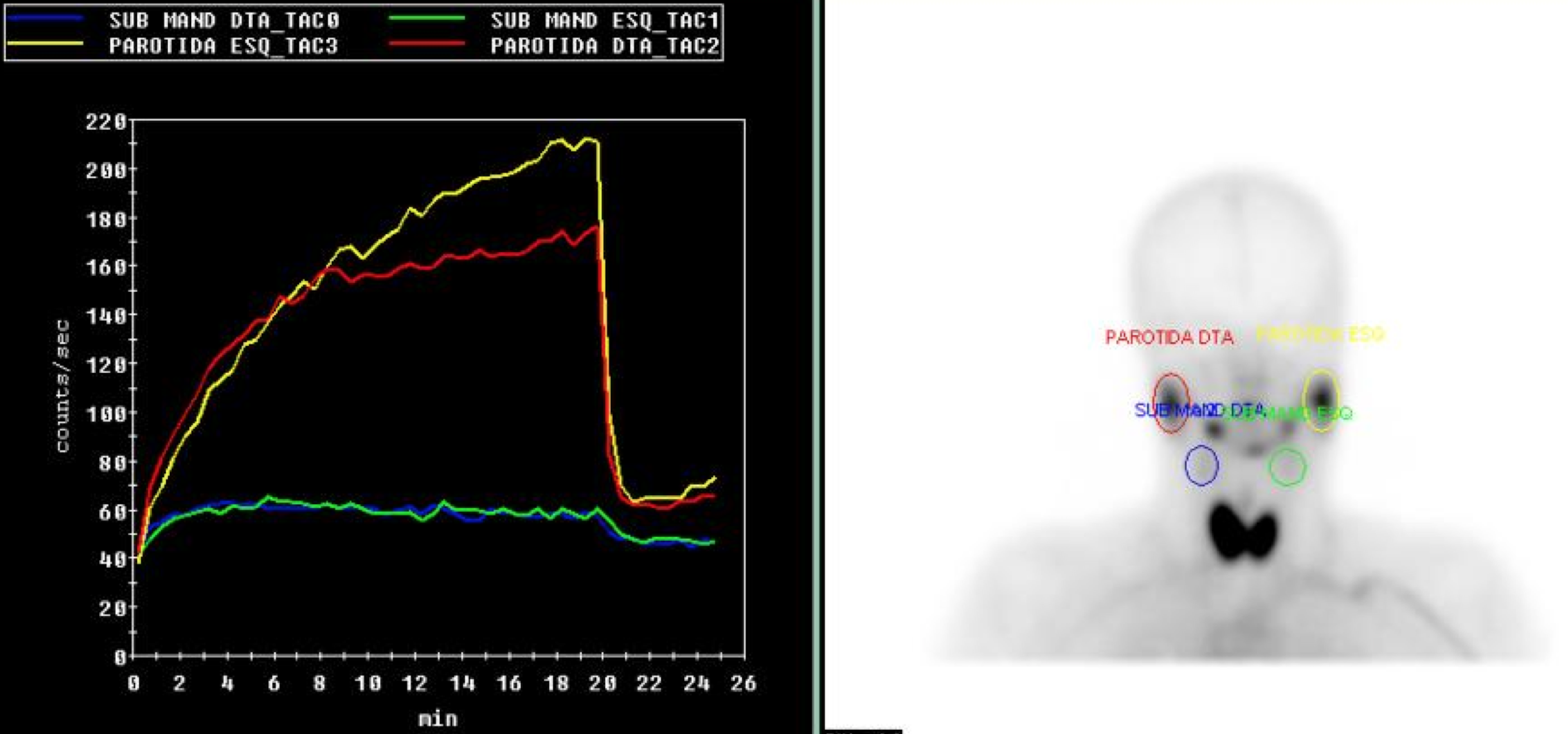
Salivary gland scintigraphy showing no trace uptake by submandibular glands
Figura IV
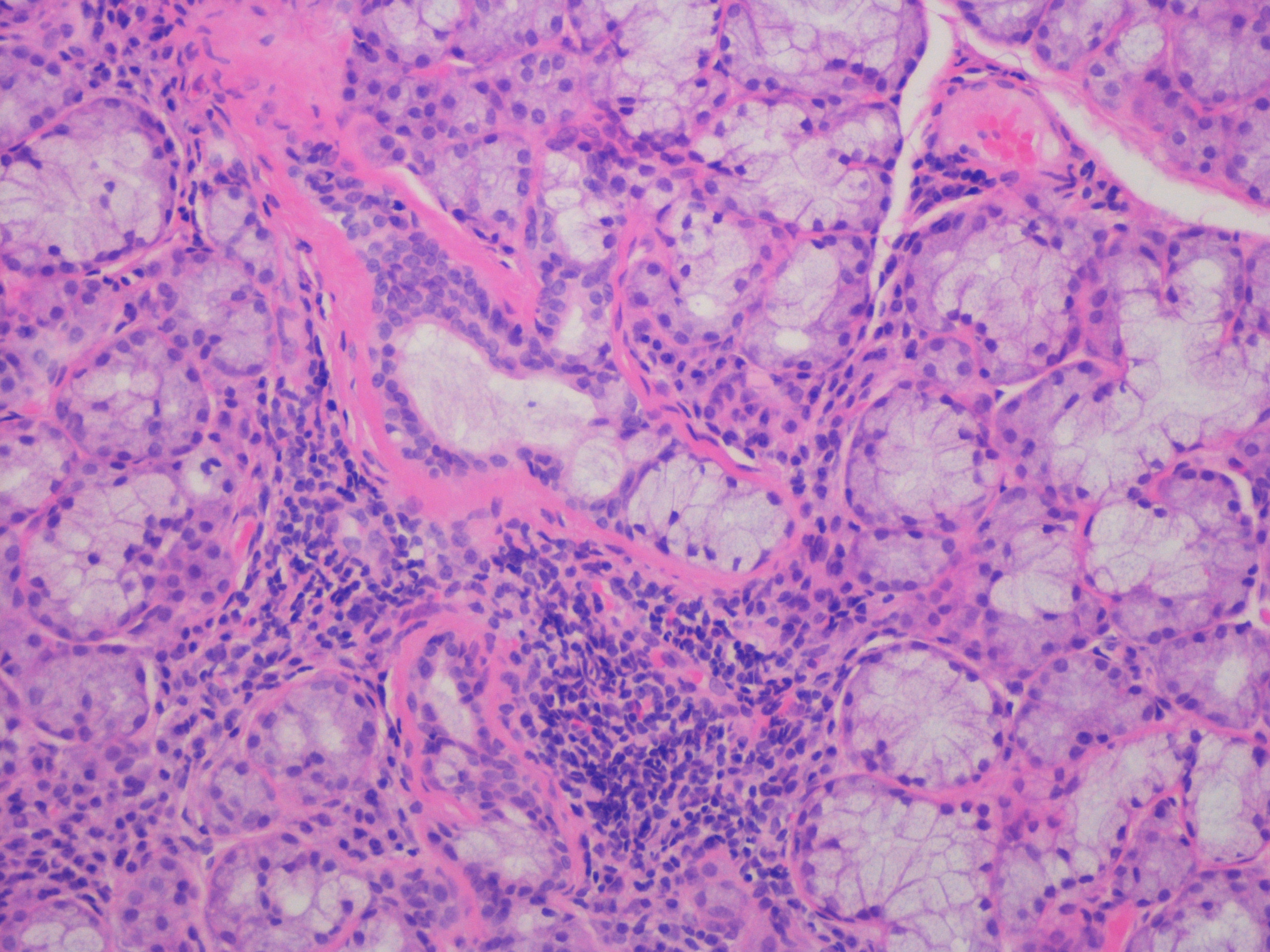
Histology of the labial gland shows focal lymphocytic sialadenitis, with a focus score 1 focus/4mm2(hematoxylin and eosin staining; magnification x40)
BIBLIOGRAFIA
1. Brito-Zeron P, Theander E, Baldini C, Seror R, Retamozo S, Quartuccio L, et al. Early diagnosis of primary Sjogren´s syndrome: EULAR-SS task force clinical recommendations. Expert Rev Clin Immunol. 2016;12(2):137-56.
2. Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, et al. Systemic lupus erythematosus: clinical and immunologic patterns of disease expression in a cohort of 1,000 patients. The European Working Party on Systemic Lupus Erythematosus. Medicine (Baltimore). 1993;72(2):113-24.
3. Costallat GL, Appenzeller S, Costallat LT. Evans syndrome and systemic lupus erythematosus: clinical presentation and outcome. Joint Bone Spine. 2012;79(4):362-4.
4. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358(9):929-39.
5. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-86.
6. Yang Y, Li Z, Wang L, Zhang F. The clinical and laboratory characteristics of Sjogren´s syndrome that progresses to systemic lupus erythematosus: a retrospective case-control study. Int J Rheum Dis. 2013;16(2):173-7.

