INTRODUÇÃO
A alcaptonúria (Online Mendelian Inheritance in Man ® - OMIM entry 203500) é uma doença autossómica recessiva rara, que afecta 1 em cada 100.000-250.000 pessoas, em igual proporção ambos os sexos 1,2. É causada pela deficiência da 1,2-Dioxigenase do homogentisato, uma enzima produzida maioritariamente no fígado, integrante do metabolismo da tirosina1 (Fig. 1). Como consequência, existe um aumento sérico e urinário de ácido homogentísico (AHG) e a polimerização dos produtos de oxidação do AHG leva à sua deposição sob a forma de um pigmento semelhante à melanina no tecido conjuntivo 1,3,4. Descreve-se um caso típico de alcaptonúria com as mais frequentes manifestações inerentes ao diagnóstico.
CASO CLÍNICO
Apresenta-se um homem caucasiano que aos 63 anos observado em consulta de Reumatologia e Medicina Interna por raquialgias, com antecedentes de hipertensão arterial, diabetes mellitus tipo 2, obesidade e estenose aórtica moderada. Sem história familiar de alcaptonúria ou outras doenças metabólicas. As queixas de raquialgia dorsal e lombar tinham vários anos de duração, características mecânicas e episódios de agudização associados a grande limitação funcional.
Ao exame objetivo apresentava pigmentação ocronótica da pela e das escleróticas (Fig. 2), sopro holossistólico aórtico, acentuada limitação da mobilidade global da coluna, mais marcado a nível dorsal e lombar, com um teste de Schöber de 2 cm. Sem atingimento das sacroilíacas e sem evidência de mielopatia ou radiculopatia. Edema e deformação óssea dos joelhos, sem sinais inflamatórios e discreta limitação da flexão, sugestivo de gonartrose. O restante exame físico era normal, nomeadamente a nível dos ombros.
O estudo analítico era normal, nomeadamente ionograma, cálcio e fósforo, função renal e hepática e parâmetros inflamatórios. As radiografias mostraram calcificações discais múltiplas e estreitamento acentuado dos espaços intervertebrais nos segmentos dorsal e lombar (Fig. 3).
A semelhança a uma espondilite anquilosante em associação com pigmentação das escleróticas, juntamente com as alterações radiológicas, levaram à suspeita de uma artropatia ocronótica. Realizou o doseamento do AHG na urina de 24 horas que confirmou o diagnóstico de Alcaptonúria. Não foi efectuado estudo genético. Foi medicado com analgésicos, anti-inflamatórios e fisioterapia.
Durante o seguimento, o doente apresentou gonalgia direita de agravamento progressivo, com gonartrose do compartimento interno com valgo do joelho na radiografia. Por esse motivo, com 65 anos de idade, foi proposto para colocação de prótese total do joelho, que não realizou por elevado risco anestésico associado a uma estenose aórtica severa. Paralelamente, foi proposto para cirurgia de substituição valvular aórtica mas rejeitou.
No decorrer da vigilância ocorreu um agravamento clínico até que aos 71 anos, foi internamento por insuficiência cardíaca descompensada pela estenose aórtica severa, com um ecocardiograma com um gradiente transvalvular máximo de 105mmHg, médio de 59mmHg, e uma área funcional de 0,5 cm2. Nesta fase, aceitou ser submetido a cirurgia de substituição valvular com colocação de uma prótese biológica. Durante o procedimento que decorreu sem intercorrências, observada a coloração escura da aorta e dos folhetos da válvula aórtica.
Posteriormente, manteve razoável qualidade de vida. Não apresentando até à data outras manifestações da doença nomeadamente nefrolitíase.
Os 2 filhos do doente não apresentavam alterações sugestivas de alcaptonúria tendo sido recomendada vigilância.
DISCUSSÃO
A alcaptonúria é uma doença icónica por ser uma das primeiras a demonstrar uma hereditariedade mendeliana recessiva, descrita por Archibald Garrod em 1902 1,3. As doenças hereditárias do metabolismo associam-se à idade pediátrica mas um número crescente é diagnosticado na idade adulta, sendo a alcaptonúria um bom exemplo disso. A única alteração a ocorrer desde a nascença é o escurecimento da urina após repousar ao ar 1. Cerca da terceira década de vida, desenvolve-se a ocronose, uma pigmentação preto-azulada, mais frequentemente nas escleróticas e pavilhões auriculares mas que também ocorrer na pele ou em fluídos corporais como o suor 1. A artropatia ocronótica inicia-se perto dos 50 anos e afeta inicialmente a coluna, causando lombalgia, rigidez articular e perda de altura 3. Ocorre um estreitamento dos espaços intervertebrais e múltiplas e exuberantes calcificações discais 3,5. Assimila-se nalguns casos a uma osteoartrose precoce ou a uma espondilartrite axial 1,3,5,6. Alguns anos mais tarde, esta artropatia severa envolve também as grandes articulações como joelho, anca e ombro 1,3,6.
Estas três manifestações são geralmente consideradas as três fases da doença, contudo a alcaptonúria também pode afetar outros sistemas 3. As complicações cardiovasculares ocorrem em idades mais avançadas 1. Há deposição no endocárdio, aorta, válvulas cardíacas e artérias coronárias 1,3. A estenose aórtica é a manifestação cardíaca mais frequente, atingindo 17% dos doentes 7,8. A nível renal, a alcaptonúria pode originar cálculos renais e prostáticos e algum grau de insuficiência renal por deposição a nível das células glomerulares e destruição do tecido conjuntivo. Outras manifestações incluem rutura de tendões e ligamentos, osteoporose e fraturas 1.
O diagnóstico implica uma elevada suspeição clínica e baseia-se na deteção de níveis elevados de AHG numa amostra de urina.
Quanto ao prognóstico, a esperança de vida não é afetada, mas associa-se a significativa morbilidade com redução da qualidade de vida 1,2.
O tratamento é paliativo e centra-se no controlo sintomático com analgesia, na fisioterapia e nalguns casos cirurgia, seja ela ortopédica ou cardíaca 1. Isto é fundamental para aumentar a qualidade de vida dos doentes com alcaptonúria. Dos estudos realizados com o intuito de reduzir a formação do AHG (vitamina C, acetilcisteína, dieta hipoproteíca, nitisinona) nenhum demostrou alterar a progressão da doença 1,4. A nitisinona é a que apresenta melhores resultados bioquímicos mas sem tradução na evolução do atingimento osteoarticular e está associada a efeitos secundários nomeadamente deposição de cristais na córnea pelo bloqueio a montante no metabolismo da tirosina 4,9. Ainda não existe terapêutica de substituição enzimática e o transplante hepático não é adequado para o tratamento da doença dada a normal esperança de vida 1,4. Para o desenvolvimento de terapêuticas futuras é necessário primeiro um melhor conhecimento da história natural da doença 10.
Em conclusão, o caso apresentado representa a evolução típica de um doente com alcaptonúria. A alcaptonúria é um bom exemplo do novo paradigma das doenças hereditárias do metabolismo, com um número crescente de diagnósticos em idade adulta.
Figura I
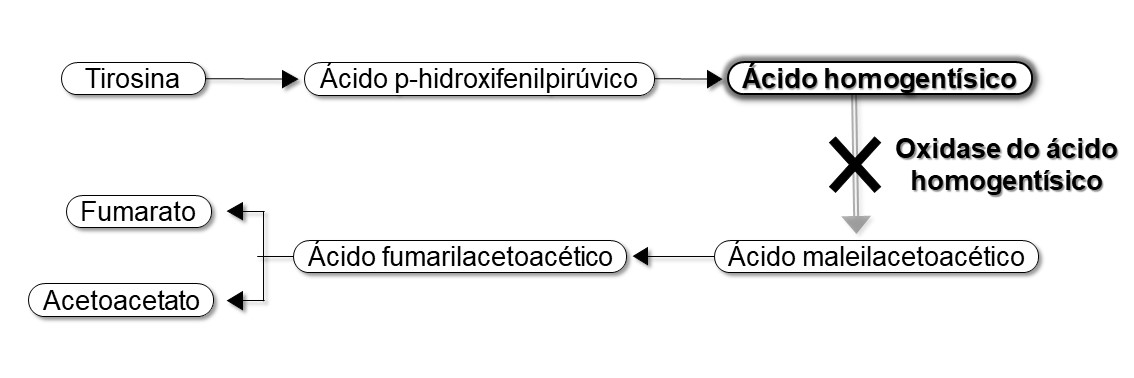
Metabolismo de degradação da tirosina. Na alcaptonúria não há oxidase do ácido homogentísico havendo acumulação de ácido homogentísico
Figura II
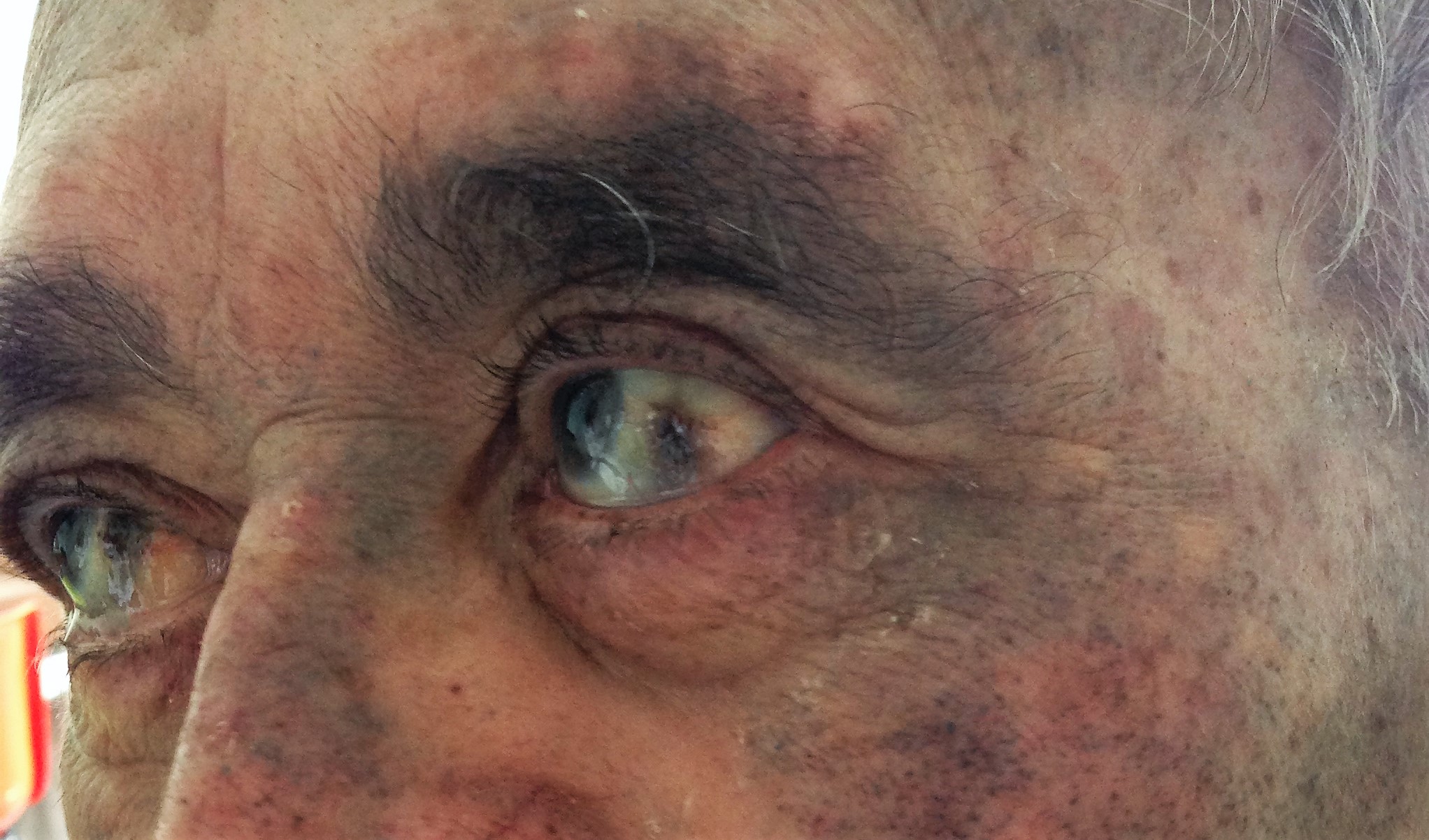
Ocronose das escleróticas e da pele
Figura III
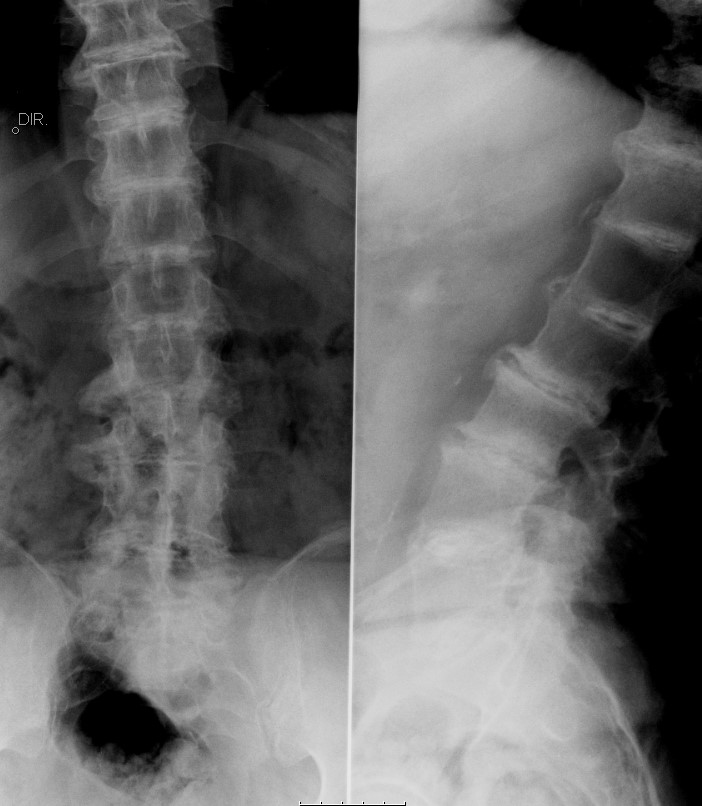
Radiografia da coluna lombar de face e perfil. Observam-se calcificações discais múltiplas com osteofitose exuberante e estreitamento acentuado dos espaços intervertebrais
BIBLIOGRAFIA
1) Mistry J, Bukhari M, Taylor A. Alkaptonuria. Rare Dis. 2013;1:1-7
2) Damarla N, Linga P, Goyal M, Tadisina S, Reddy S, Bommisetti H. Alkaptonuria: A case report. Indian J Ophthalmol. 2017;65: 518521.
3) Aquaron R. Alkaptonuria: A Very Rare Metabolic Disorder. Indian J Biochem Biophys. 2013;50:339-344
4) Arnoux J, Sang K, Brassier A, Grisel C, Servais A, Wippf J, et al. Old treatments for new insights and strategies: proposed management in adults and children with alkaptonuria. J Inherit Metab Dis. 2015;38:791-796
5) Lok Z, Goldstein J, Smith J. Alkaptonuria-Associated Aortic Stenosis. J Card Surg 2013;28:417420
6) Hannoush H, Introne W, Chen M, Lee S, O´Brien K, Suwannarat P, et al. Aortic Stenosis and Vascular Calcifications in Alkaptonuria. Mol Genet Metab. 2012;105:198202
7) Ranganath L, Jarvis J, Gallagher J. Recent advances in management of alkaptonuria(invited review; best practice article). J Clin Pathol. 2013;66:367373
8) Ranganath L, Timmis O, Gallagher J. Progress in Alkaptonuria are we near to an effective therapy? J Inherit Metab Dis. 2015;38:787789.
9- Introne W, Perry M, Troendle J, Tsilou E, Kayser M, Suwannarat P et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314
10- Ranganath L, Cox T. Natural history of alkaptonuria revisited: analyses based on scoring systems. J Inherit Metab Dis. 2011;34:11411151

