Introdução:
A doença de McArdle ou Glicogenose Tipo V (OMIM #232600) é uma patologia metabólica hereditária que afeta apenas o músculo esquelético, com um padrão de transmissão autossómico recessivo(1).
Manifesta-se habitualmente por mialgias e fadiga rápida, que se instalam nos primeiros minutos de atividade física e que pode evoluir para quadro de rabdomiólise, com mioglobinúria e por vezes insuficiência renal aguda(2).
Serve este artigo para apresentar o caso de um doente de 27 anos, cujas queixas e achados analíticos fizeram levantar a suspeita de miopatia, que se veio a revelar uma Glicogenose Tipo V.
Caso Clínico:
Doente de 27 anos, género masculino, é referenciado ao Serviço de Urgência pelo seu Médico de Família por elevação persistente da creatinofosfocinase (CPK) e da mioglobina. Ao inquérito sintomático manifestava queixas de mialgias generalizadas sem predomínio pelos grupos musculares proximais ou distais, astenia marcada e fadiga muscular fácil, com limitação importante nas atividades de vida diária. Estas queixas tinham um ano de evolução, associadas a períodos de exercício isométrico realizados no contexto laboral, surgindo nos primeiros minutos do esforço e melhorando com o repouso. Não havia história de exercício físico violento realizado recentemente e ao inquérito, o doente negava que as queixas melhorassem se retomasse a atividade física após um breve momento de repouso. Negava história de alterações da cor urinária no momento da observação ou prévias. Não existiam antecedentes pessoais de relevo. Referia apenas história familiar de dislipidemia e diabetes mellitus, sem história conhecida de doenças neuromusculares na família. Não existia história de hábitos medicamentosos ou de utilização de substâncias de abuso. Ao exame objetivo, salientavam-se a ausência de défices na força muscular, reflexos miotáticos presentes, normais e simétricos e a ausência de fasciculações. Analiticamente, apresentava como alterações de relevo uma elevação da CPK de cerca de vinte vezes em relação ao valor normal (CPK 26818U/L), elevação da LDH de 1521U/L e doseamento de mioglobina na urina negativo. Do estudo da autoimunidade o doente apresentava anticorpos antinucleares e anti-DNAds com títulos negativos. Não existia rebate sobre a função renal. Nesta altura, foi internado para esclarecimento etiológico do quadro de rabdomiólise.
Durante o internamento o doente foi submetido a hidratação endovenosa abundante, com normalização progressiva da CPK e da LDH. Realizou estudo por eletromiografia com estimulação repetitiva sem alterações, e teve alta ao fim de oito dias, clínica e analiticamente melhorado, com indicação para evicção de exercício físico intenso.
Foi orientado para Consulta Externa de Neurologia, com agendamento de biópsia muscular, que revelou presença ocasional de fibras necrosadas (ghost fibers), fibras com vacuolizações arredondadas, múltiplas e subsarcolémicas, com material PAS positivo e ausência de atividade miofosforilásica (Figura 1). Estabeleceu-se nesta altura o diagnóstico de Doença de McArdle.
Desde então o doente apresentou melhoria das mialgias, mantendo queixas de astenia. Apresenta valores oscilantes de CPK, que se correlacionam com períodos de maior atividade física e com a má adesão à terapêutica proposta, nomeadamente à ingestão de dieta rica em hidratos de carbono complexos e de açucares de absorção rápida em antecipação a períodos de maior exigência física.
Discussão:
A doença de McArdle ou Glicogenose Tipo V é uma patologia metabólica hereditária que afeta apenas o músculo esquelético(1). Tem padrão de transmissão autossómico recessivo, e é causada pela presença de mutações em ambos os alelos do gene PYGM, que codifica a miofosforilase muscular, condicionando um défice absoluto de atividade desta enzima no músculo, mas poupando a expressão das isoformas hepática e cerebral. O gene PYGM localiza-se no cromossoma 11q13, e são múltiplas as mutações associadas a esta patologia não tendo sido estabelecida até à data qualquer relação entre mutações específicas e o grau de severidade da doença(3). A miofosforilase é responsável pela libertação de glicose das reservas musculares de glicogénio, que será a fonte principal de energia utilizada em eventos de exercício físico muito intenso e de exercício físico em condições de anaerobiose(4). É uma doença rara, para a qual existem poucos estudos epidemiológicos que afiram a sua real incidência(2)(3).
A clínica pode ser altamente variável, com uma idade de apresentação e tolerância ao esforço que podem diferir bastante de caso para caso(5). Esta variabilidade pode ser explicada pela biodisponibilidade de fontes energéticas alternativas, pela dieta e estilo de vida adotados por cada pessoa, estando descrita uma mutação no gene da Enzima de Conversão da Angiotensina relacionada com perfil fenotípico mais adverso(6). Os primeiros sintomas surgem habitualmente na segunda e terceira décadas de vida, com episódios de mialgias e fadiga que surgem rapidamente após o início de exercício físico isométrico ou aeróbio mantido, sendo que em alguns casos, um breve período de repouso permite a resolução da clínica e o retomar da atividade física sem queixas (second wing phenomenon). Se o exercício físico for mantido apesar da sintomatologia, irá ocorrer rabdomiólise e contractura muscular, com quadros de mioglobinúria recorrente, descritos nalguns doentes(2).
Analiticamente, a alteração mais frequente é uma elevação importante e persistente da CPK, mesmo em repouso(2)(5). O diagnóstico definitivo é feito pelo estudo genético, com demonstração de mutações associadas à doença, ou por biópsia muscular com posterior método imunohistoquímico, que demonstra a ausência de atividade da miofosforilase(3).
Neste doente, a sintomatologia apontava para processo miopático, tendo sido colocadas várias hipóteses diagnósticas. A hipótese de miopatia inflamatória, embora considerada pouco provável, dado que a clínica assentava sobretudo em mialgias (que não dominam o quadro clínico das miopatias inflamatórias) e dada a ausência de alterações na força muscular, era no entanto, a hipótese mais preocupante e o principal motivo que levou à realização da biópsia muscular. A hipótese de miopatia metabólica era considerada bastante provável, atendendo às manifestações desencadeadas pelo esforço físico e à ausência de défices na força muscular. Apesar de tudo, os episódios de mioglobinúria, manifestação clássica deste grupo de patologias, nunca foi reportado pelo nosso doente. Apesar de não existir história medicamentosa ou de exposições ambientais, a hipótese de miopatia de causa tóxica foi outra hipótese levantada e que foi tida em consideração. Neste caso, a biópsia muscular acabou por fornecer o diagnóstico definitivo.
O tratamento é apenas sintomático, existindo apenas estudos com pequeno número de participantes. A utilização de baixas doses de creatina (60mg/kg/dia), a suplementação com sucrose oral, imediatamente antes de períodos de exercício físico, a adoção de dieta rica em hidratos de carbono e a manutenção de exercício físico aeróbico regular de intensidade moderada parecem demonstrar benefícios em termos de tolerância ao esforço e de diminuição da sintomatologia(6).
Conclusão:
A doença de McArdle é uma miopatia metabólica rara, que afeta o músculo esquelético e que se manifesta com queixas de mialgias e fadiga rápida, que se instala nos primeiros minutos de atividade física e que pode evoluir para quadro de rabdomiólise, com mioglobinúria, cursando por vezes com insuficiência renal aguda. No caso apresentado, a sintomatologia era compatível com esta patologia, bem como a elevação persistente da CPK. À data do internamento, o doente apresentava um quadro de rabdomiólise importante, embora nunca tenha descrito um quadro compatível com mioglobinúria. O tratamento é apenas sintomático assenta essencialmente na adoção de alterações dietéticas, com dieta rica em hidratos de carbono, e de estilo de vida, com prática regular de exercício físico de moderada intensidade. Neste caso, a má adesão às recomendações fornecidas dificulta o controlo sintomático do doente, que mantém períodos de mialgias importantes, com impacto na sua qualidade de vida.
Figura I
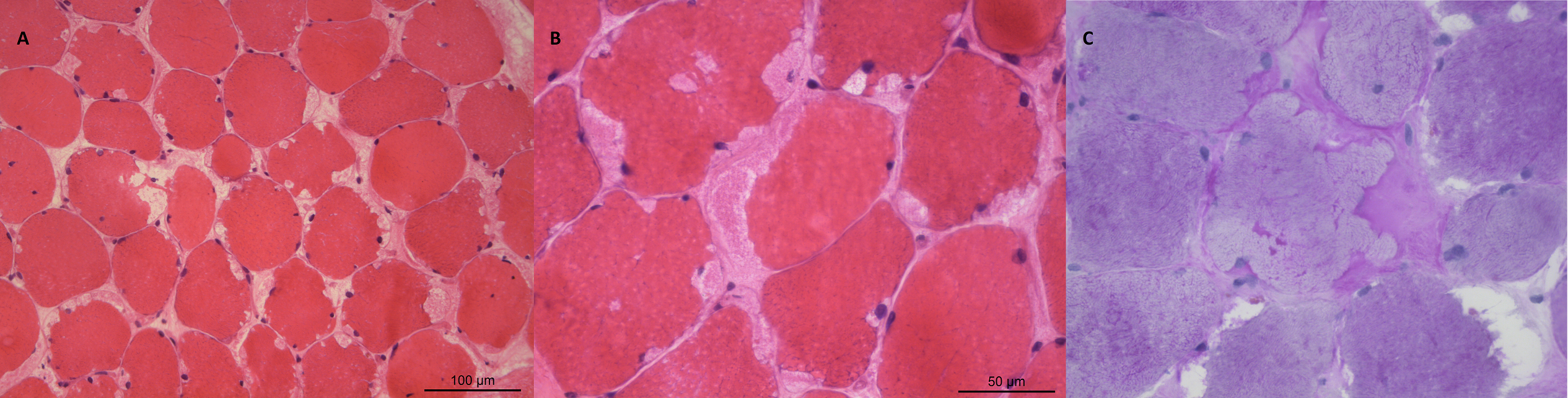
Imagens histológicas da biópsia muscular; A - Coloração com hematoxilina e eosina (HE), pequena ampliação. Imagem evidenciando fibras musculares atróficas e vacuolizações subsarcolémicas; B - Coloração com HE, grande ampliação. Imagem evidenciando vacuolizações arredondas, múltiplas, subsarcolémicas; C Coloração com ácido periódico-Schiff (PAS), grande ampliação. Imagem evidenciando vacuolizações subsarcolémicas contendo material PAS positico (glicogénio).
BIBLIOGRAFIA
1. Scalco RS, Morrow JM, Booth S, Chatfield S, Godfrey R, Quinlivan R. Misdiagnosis is an important factor for diagnostic delay in McArdle disease. Neuromuscul Disord [Internet]. 2017;27(9):8525. Available from: http://dx.doi.org/10.1016/j.nmd.2017.04.013
2. Santalla A, Nogales-Gadea G, Encinar AB, Vieitez I, González-Quintana A, Serrano-Lorenzo P, et al. Genotypic and phenotypic features of all Spanish patients with McArdle disease: A 2016 update. BMC Genomics. 2017;18(Suppl 8).
3. De Castro M, Johnston J, Biesecker L. Determining the prevalence of McArdle disease from gene frequency by analysis of next-generation sequencing data. Genet Med. 2015;17(12):10026.
4. Nogales-Gadea G, Godfrey R, Santalla A, Coll-Cantí J, Pintos-Morell G, Pinós T, et al. Genes and exercise intolerance: insights from McArdle disease. Physiol Genomics [Internet]. 2016;48(2):93100. Available from: http://physiolgenomics.physiology.org/lookup/doi/10.1152/physiolgenomics.00076.2015
5. Lorenzoni, P.J., Werneck, L.C., Kay, C.S.K. et al. Single-centre experience on genotypic and phenotypic features of southern Brazilian patients with McArdle disease. Acta Neurol Belg 2018.
6. Quinlivan R, Rj B. Pharmacological and nutritional treatment for McArdle s disease ( Glycogen Storage Disease type V ) ( Review ). 2005;(1).

