INTRODUÇÃO
Os tumores neuroendócrinos (NET) do pâncreas são neoplasias raras, com incidência de 0,48 por 100 000 pessoas, podendo produzir variados peptídeos hormonais, resultando numa variedade de síndromes clínicos. A maioria destas neoplasias são não-funcionantes, como tal não associados a síndrome clínico.1 Dos tumores funcionantes, os insulinomas são os mais comuns (70%) seguidos dos glucagonomas (15%), gastrinomas e somatostinomas (10%). Os restantes incluem os VIPomas, ACTHomas, GRFomas, tumores produtores de calcitonina ou de peptídeo relacionado com a hormona paratiroide, entre outros.2
Os ACTHomas, localizados no pâncreas, produzem ACTH (Adrenocorticotropic hormone) sendo responsáveis por 4 a 16% dos casos de Síndrome de Cushing ectópico (SCE).3
As manifestações do SCE dependem do tecido de origem e do tipo de tumor que segrega a ACTH. Em geral, os doentes apresentam-se com fraqueza e atrofia musculares por miopatia proximal, pele fina e equimoses fáceis, hipertensão, alteração do metabolismo dos lípidos, hiperpigmentação e alterações psiquiátricas como psicose e depressão.4 A produção excessiva de cortisol (dependente da secreção ectópica de ACTH) contribui para o efeito de excesso de mineralocorticoide, resultando na excreção renal de potássio e hidrogenião e consequente hipocalémia e alcalose metabólica graves.5, 6
CASO CLÍNICO
Mulher de 72 anos, autónoma, sem antecedentes patológicos de relevo e sem medicação regular.
No mês prévio à admissão, desenvolveu lombociatalgia direita aliviada com repouso e medicação (toma única de betametasona intramuscular, glucosamina e suplementos de cálcio). Duas semanas depois, por surgirem astenia, edema facial, lacrimejo, xerostomia e eritema do pescoço e do tronco suspendeu a referida medicação. Por persistência do edema facial, foi medicada três dias antes com furosemida (fez apenas 40mg num dia e 80mg no seguinte), tendo desenvolvido quadro de fraqueza nos membros inferiores e desequilíbrio, pelo que recorreu ao Serviço de Urgência. Na admissão apresentava pressão arterial 150/83 mmHg, edema facial, eritema macular, não pruriginoso da face anterior e posterior do tronco, com desaparecimento à digitopressão, e edema maleolar. Ao exame neurológico documentou-se fraqueza muscular na cintura pélvica (manobra de Gowers positiva), sem fasciculações ou fatigabilidade. O restante exame objetivo não apresentava alterações.
Do estudo efetuado (Tabela 1) salientava-se alcalose metabólica (pH 7,68 e HCO3 superior a 60 mmol/L), glicemia ocasional 200mg/dL, hipocalémia (2,0 mmol/L) e hipercalúria (70,6 mmol/L).
No internamento no Serviço de Medicina Interna foi continuada a investigação do quadro, verificando-se: níveis de aldosterona, renina e relação aldosterona/ renina normais; ACTH e cortisol (avaliado pelo método de quimioluminescência) sérico e urinário aumentados (Tabela 2). No teste de supressão com dexametasona em alta dose (8 mg de dexametasona pelas 24h da véspera e avaliação de cortisol e ACTH séricos pelas 8h) (Tabela 2), manteve níveis de ACTH e cortisol sérico aumentados, sugerindo secreção ectópica de ACTH.
A ressonância magnética (RM) mostrou hipófise de morfologia normal (Fig. 1) e a tomografia computorizada (TC) cervico-tóraco-abdominal (sem contraste, por alergia ao iodo) não apresentou alterações (Fig. 2).
Na cintigrafia com octreótido (Fig. 3) verificou-se um foco de captação anómala na cavidade abdominal, anteriormente ao rim direito (em topografia compatível com a cabeça do pâncreas) sugestiva de lesão tumoral com hiperexpressão anómala de recetores de somatostatina (provável lesão produtora de ACTH). A RM abdominal mostrou um pâncreas de características normais, sem evidência de lesões expansivas. Os doseamentos de serotonina, cromogranina A e ácido 5-hidroxindolacético urinário (Tabela 2) foram normais.
Durante o internamento, sem toma de diurético, a doente necessitou de suplementação de potássio (intravenoso e posteriormente oral), espironolactona e acetazolamida para controlo dos distúrbios eletrolíticos e ácido-base.
A ocorrência de complicações (peritonite secundária a diverticulite aguda necessitando de cirurgia urgente e pós-operatório em Unidade de Cuidados Intensivos, deiscência e infeção da ferida cirúrgica e pneumonia nosocomial) impediram a continuação da marcha diagnóstica, nomeadamente a realização de tomografia com emissão de positrões com peptídeo análogo da somatostatina (PET DOTA-NOC) para melhor esclarecimento e avaliação da extensão da lesão. A doente veio a falecer no contexto da infeção respiratória.
DISCUSSÃO
Na presença de alcalose metabólica, hipocalémia inexplicada e hipertensão arterial deve pensar-se em excesso primário de mineralocorticoide. As duas outras causas major de hipertensão e hipocalémia são a doença renovascular e o tratamento com diurético. Nas causas menos comuns inclui-se o Síndrome de Cushing.7A avaliação inicial passa pelo doseamento de aldosterona e renina. Associadamente, e tendo em conta a presença de edema facial e eritema do tronco (apresentados pela doente em análise), a presença de hipercortisolismo deve igualmente ser investigada através do doseamento de cortisol urinário ou salivar noturno e do teste de supressão com dexametasona. Após a confirmação de hipercortisolismo há que identificar a causa, através do doseamento de ACTH sérico e realização de exame de imagem dirigido à glândula supra-renal.4
A betametasona intramuscular apresenta uma semi-vida plasmática de aproximadamente 5h, ocorrendo redução dos níveis séricos de cortisol, mas com recuperação para níveis basais após 4h.8 Também a excreção urinária de cortisol pode ser alterada, mas apenas até 19 dias após a toma. Assim, tendo os sintomas da doente surgido 15 dias após a toma desse fármaco e a recorrência ao hospital ter sido 40 dias após, não é expectável que a toma prévia de betametasona possa ter interferência nos achados laboratoriais do caso em análise.8
Na presença de hipercortisolismo dependente de ACTH com glândula suprarrenal imagiologicamente de características normais, a hipótese diagnóstica mais provável torna-se um tumor produtor de ACTH. A exclusão de fonte hipofisária de ACTH torna-se importante já que a grande maioria dos casos é devido a adenoma corticotrófico da hipófise. No entanto, cerca de 10 a 15% das situações é devida a secreção ectópica de ACTH por um tumor não hipofisário.8, 9 A RM é a técnica de imagem mais sensível para a deteção destes tumores. Contudo, identifica apenas 50% dos adenomas da hipófise, já que a sua maioria são microadenomas (inferiores a 6mm)10, devendo-se por isso recorrer também a outros métodos não invasivos como a prova de supressão com dexametasona em alta dose e/ou o teste de estimulação com Corticotropin releasing hormone (CRH).11
A ausência de supressão (ACTH >20 pg/mL) no teste com dexametasona em alta dose confirma a existência de fonte ectópica de produção de ACTH, sendo responsável por cerca de 15-20% dos casos de Síndrome de Cushing dependente de ACTH.11 A maioria destes casos é causada por carcinomas do pulmão, pâncreas ou timo, sendo que, destes, o mais comum (cerca de 50%) é o carcinoma pulmonar de pequenas células.4
A melhor abordagem para identificação da neoplasia primária pode ser difícil, pelo que a TC e a RM, complementadas com a cintigrafia com 111-In-octreótido (Octreoscan), são úteis para o diagnóstico. O Octreosan baseia-se na presença de recetores da somatostatina nas células tumorais com diferenciação neuroendócrina.4, 10
No caso em análise, não foi possível identificar a localização do tumor na TC nem na RM, o que é concordante com o descrito na literatura - 8 a 50% dos doentes sem identificação em TC ou RM da fonte da secreção de ACTH.4 Neste caso a localização tornou-se clara apenas na cintigrafia com octreótido. É de salientar que, por alergia, as TC efetuadas nesta doente foram sem contraste, o que poderá ter dificultado o diagnóstico, já que muitos dos NET do pâncreas são hipervascularizados, tornando as TC contrastadas altamente precisas.12 Em determinadas circunstâncias, como no caso exposto, em que há suspeita de um NET funcionante e os exames solicitados (TC, RM e cintigrafia com Octreótido) são negativos, a PET DOTA-NOC é um exame com maior sensibilidade e especificidade.
O exame histológico é essencial para confirmar o diagnóstico e definir o melhor tratamento. Apesar de o grupo dos NET gastro-entero-pancreáticos ser heterogéneo, todos partilham um fenótipo comum com imunorreatividade para os marcadores cromogranina A e sinaptofisina. Uma descrição dos achados macro e microscópicos e das características imunohistoquímicas é mandatória para a correta classificação e estadiamento.2, 13
O tratamento destas neoplasias é frequentemente multimodal. Quando a condição clínica do doente o permite, a cirurgia com intuito curativo deve ser considerada, mesmo na presença de doença metastizada. O esvaziamento ganglionar regional é mandatório.2, 13
Os análogos da somatostatina (octreótideo, lanreótido) são o tratamento sistémico standard nos NET funcionantes de qualquer localização e nos NET não funcionantes bem ou moderadamente diferenciados. A sua eficácia anti-tumoral parece fraca no que respeita a resposta tumoral objetiva (5-10%); no entanto, a taxa de estabilização tumoral pode chegar aos 50-60%. Nos tumores indiferenciados, esta abordagem terapêutica não está recomendada. O everolimus e os inibidores da tirosina cinase (sunitinib e pazopanib) demonstraram eficácia anti-tumoral nos NET pancreáticos.2, 13
Apesar dos avanços terapêuticos, os NET produtores de ACTH exibem comportamento agressivo, com elevadas taxas de metastização (88% em fases iniciais) e de mortalidade (superior a 60% aos 24 meses).14
Além da história natural destes tumores, a imunossupressão associada à hipercortisolemia aumenta a prevalência de infeções e outras patologias agudas (pancreatite, inflamação intestinal com perfuração e peritonite, meningite, insuficiência cardíaca e embolia pulmonar), sendo também estas causas de morte prevalentes.4, 15 Assim, torna-se fundamental o controlo rápido da hipercortisolemia, seja por meio farmacológico (análogos da somatostatina e quimioterapia) ou cirúrgico (remoção do tumor primário).15
No caso exposto, o diagnóstico histológico não foi possível, limitando a implementação de um tratamento direcionado.
CONCLUSÃO
Apresenta-se uma situação clínica rara que ilustra a dificuldade na marcha diagnóstica dos NET do pâncreas, a sua associação a outras patologias e o mau prognóstico. É fundamental a suspeita clínica que despolete uma intervenção diagnóstica e terapêutica atempadas.
Figura I
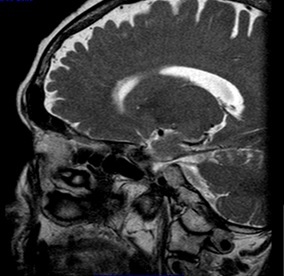
Ressonância magnética hipofisária hipófise de morfologia normal e sinal homogéneo
Figura II
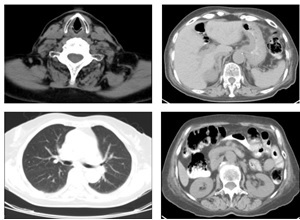
Tomografia computorizada cervico-toraco-abdomino-pélvica sem massas ou outros aspetos patológicos no compartimento cervical e torácico. Abdómen e pelve sem alterações tomodensitométricas aparentes
Figura III
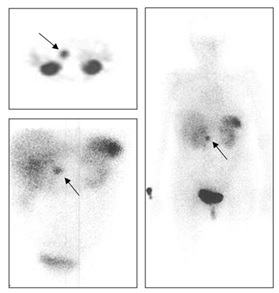
Cintigrafia com octreótido - foco de captação anómala na cavidade abdominal, anteriormente ao rim direito (na topografia da cabeça do pâncreas) sugestiva de lesão tumoral com hiperexpressão anómala de recetores de somatostatina.
Figura IV
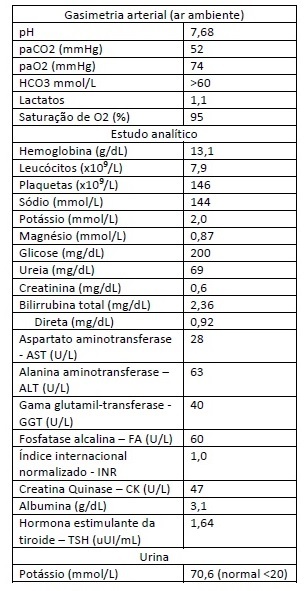
Tabela 1 - Valores laboratoriais na admissão
Figura V
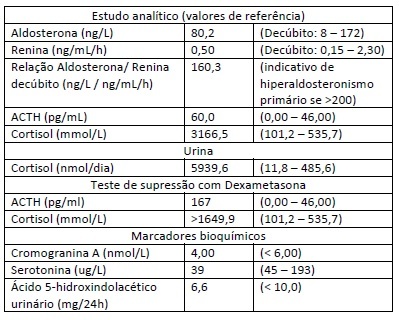
Tabela 2 - Estudo etiológico laboratorial
BIBLIOGRAFIA
1. Dasari A, Shen C, Halperin D, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol 2017;3:1335-42.
2. Shah M, Goldner W, Halfdanarson T, Bergsland E, Berlin J, Halperin D, et al. NCCN Guidelines® Insights Neuroendocrine and Adrenal Tumors, Version 2.2018. J Natl Compr Canc Netw. 2018;16(6):693702
3. Jensen R, Cadiot G, Brandi M, Herder W, Kaltsas G, Komminoth P, et al. ENETS Consensus Guidelines of the Management of Patients with Digestive Neuroendocrine Neoplasms: Functional Pancreatic Endocrine Tumor Syndromes. Neuroendocrinology 2012; 95: 98-119
4. Alexandraki KI, Grossman AB. The ectopic ACTH syndrome. Rev Endocr Metab Disord. 2010. Jun;11(2):117-26
5. Ulick S, Wang JZ, Blumenfeld JD, Pickering TG. Cortisol inactivation overload: a mechanism of mineralocorticoid hypertension in the ectopic adrenocorticotropin syndrome. J Clin Endocrinol Metab. 1992;74(5):963.
6. Torpy DJ, Mullen N, Ilias I, Nieman LK. Association of hypertension and hypokalemia with Cushing´s syndrome caused by ectopic ACTH secretion: a series of 58 cases. Ann N Y Acad Sci. 2002;970:134.
7. Fauci AS, Braunwald E, Kasper DL, Hauser SL, Longo DL, Jameson JL, et al. Harrison´s Principles of Internal Medicine. Mc Graw Hill Medical, 17ª edição, p. 2212-2214 e 2253-2259
8. Infarmed. Diprofos Depot Suspensão injetável: resumo das características do medicamento. 2013
9. Woo YS, Isidori AM, Wat WZ, Kaltsas GA, Afshar F, Sabin I, et al. Clinical and biochemical characteristics of adrenocorticotropin-secreting macroadenomas. J Clin Endocrinol Metab. 2005;90(8):4963.
10. Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994;120(10):817.
11. Santos S, Santos E, Gaztambide S, Salvador J. Diagnosis and differential diagnosis of Cushing´s syndrome. Endocrinol Nutr. 2009 Feb;56(2):71-84.
12. Dromain C, de Baere T, Baudin E, Galline J, Ducreux M, Boige V, et al. MR imaging of hepatic metastases caused by neuroendocrine tumors: comparing four techniques. AJR Am J Roentgenol. 2003;180(1):121.
13. Öberg K, Knigge U, Kwekkeboom D, Perren A; ESMO Guidelines Working Group. Neuroendocrine gastro-entero-pancreatic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012 Oct;23 Suppl 7:vii124-30.
14. Clark ES, Carney JA. Pancreatic islet cell tumor associated with Cushing´s syndrome. Am J Surg Pathol 1984;8:917-924.
15. Attarian S, Libutti SK, Chuy J. ACTH-Producing Pancreatic Neuroendocrine Tumor Presenting with Severe Hypokalemic Alkalosis: A Case Report. J Gastrointest Canc. 2016 Jun;47(2):217-20.

