Introdução:
O mieloma múltiplo (MM) corresponde a 10% de todas as neoplasias hematológicas e caracteriza-se pela proliferação monoclonal de plasmócitos. Estão descritos mielomas produtores de todas as classes de cadeias pesadas e leves bem como não secretores 1,2. O subtipo produtor de IgD representa menos de 2% de todos os mielomas. Afeta indivíduos mais jovens e associa-se a mau prognóstico. Caracteriza-se por picos monoclonais pequenos ou inexistentes, predomínio de cadeias leves λ, elevada prevalência de proteinúria de Bence Jones, insuficiência renal, hipercalcémia e amiloidose 3,4.
O MM é tipicamente uma doença confinada à medula óssea e osso, contudo o subtipo IgD associa-se frequentemente a envolvimento extraósseo. A apresentação com plasmocitomas extramedulares múltiplos é bastante rara e apresenta pior prognóstico 5,6.
O tratamento recomendado inclui esquemas com bortezomib, autotransplante e terapêutica de manutenção com talidomida 7. Apesar da resposta à terapêutica ser semelhante à dos restantes subtipos, a taxa de recidiva e a sobrevida é menor nos doentes com MM IgD 8.
Tratando-se de uma doença rara existem poucos estudos randomizados e a publicação de casos clínicos continua a ser essencial.
Caso clínico:
Mulher de 64 anos, leucodérmica, sem antecedentes pessoais relevantes e sem medicação habitual. Recorreu ao Serviço de Urgência (SU) por quadro com 1 mês de evolução de dor torácica tipo pleurítica, perda de peso (6Kg em 3 meses), obstipação e diminuição da força e sensibilidade nos membros inferiores. O exame neurológico revelou força grau 2 na flexão das coxas, grau 4 na flexão e extensão das pernas e grau 5 na flexão e extensão dos pés; reflexos osteotendinosos rotulianos muito vivos com aumento da área e nível de sensibilidade álgica em D8. A ressonância magnética (RM) da coluna confirmou compressão medular dorsal por massa intra-canalar posterior a nível de D3-D6, fratura patológica de D6 e infiltração metastática de várias vértebras (Fig.1).
Face a envolvimento ósseo extenso, anemia (Hemoglobina 10.7 g/dL) e retenção azotada (ureia 79 mg/dL, creatinina 1.22 mg/dL) foi colocada a hipótese de MM apesar da presença de hipocalcémia (cálcio corrigido 88.0mg/dL). A eletroforese de proteínas mostrou pico monoclonal gama de pequenas dimensões (2.5 g/L) e a imunofixação revelou componente monoclonal IgD λ (IgD 5650mg/L, cadeias λ livres 575, λ/κ livres 9583). Ainda a referir proteinúria de Bence Jones por cadeias λ leves livres, LDH elevada (1771/uL), albumina 40.9 g/L, beta2-microglobulina 2.61 mg/L. A biópsia óssea e o mielograma não mostraram infiltração plasmocitária.
Realizou tomografia computorizada crânio-encefálica (TC-CE) que evidenciou lesão expansiva na calote frontal anterior esquerda e múltiplas lesões líticas (Figs 2 e 3). Neste contexto, considerou-se que as lesões ósseas poderiam corresponder a plasmocitomas tendo sido solicitada biópsia guiada por TC do plasmocitoma da calote craniana. No entanto, a biópsia por agulha fina apenas permitiu obtenção de escasso material, não tendo sido possível confirmar a presença de plasmocitomas.
Apesar do resultado da biópsia e após discussão com a Hematologia foi admitido o diagnóstico de MM IgD-λ com plasmocitomas extramedulares múltiplos. A doente teve alta orientada à consulta de Hematologia e iniciou quimioterapia com bortezomib, ciclofosfamida e dexametasona (CyBorD) com resposta completa ao 4º ciclo. Neste momento, encontra-se assintomática e a aguardar autotransplante medular.
Discussão:
O MM IgD é um subtipo raro de mieloma 7. A apresentação clínica é semelhante à dos restantes subtipos e inclui dor óssea, astenia e perda ponderal. Neste caso clínico, além destes a doente apresentava ainda compromisso neurológico provocado por fratura patológica de D6 e plasmocitomas extramedulares. Estes são mais frequentes no subtipo IgD e tal como nesta doente, têm origem em lesões ósseas primárias em 68-85% dos casos 6. A apresentação com plasmocitomas extramedulares múltiplos ao diagnóstico é rara estando descrita em apenas 7-18% dos casos 5.
Nesta doente o diagnóstico foi sugerido pelo envolvimento ósseo extenso e pela presença de anemia e disfunção renal. Estas são lesões de órgão alvo provocadas pela proteína M e conhecidas pelo acrónimo CRAB (hipercalcémia, disfunção renal, anemia e envolvimento ósseo) 2,9.
A eletroforese de proteínas revelou um pico monoclonal pequeno típico do subtipo IgD resultante da baixa concentração desta imunoglobulina no plasma. Tal pode atrasar ou conduzir ao diagnóstico errado de MM não secretor. A predominância de cadeias λ também é típica do MM IgD, bem como a existência de proteinúria de Bence Jones, que estão normalmente associadas à maior prevalência de disfunção renal nestes doentes 1.
O diagnóstico de MM exige além de lesões de órgão alvo a presença de ≥10% plasmócitos na biópsia óssea ou no plasmocitoma e/ou presença de biomarcadores de malignidade (≥60% plasmócitos na medula óssea, rácio cadeias leves livres ≥ 100 ou > 1 lesão focal na RM) 9. Neste caso, a doença era exclusivamente extramedular pois não existia invasão plasmocitária da medula óssea. Da mesma forma, a biópsia de plasmocitoma, pela quantidade insuficiente de material, não identificou plasmócitos. O diagnóstico foi assim presuntivo e baseado na produção monoclonal de IgD, nas lesões de órgão alvo descritas e na ausência de invasão medular típica dos mielomas com plasmocitomas extramedulares múltiplos.
O estadiamento do MM IgD é particularmente difícil pela sua raridade. O sistema de estadiamento recomendado é o Revised International Staging System (R-ISS) que combina vários fatores de prognóstico (β2-microglobulina, albumina, LDH e alterações citogenéticas). No caso apresentado o prognóstico está limitado pela impossibilidade de caracterização genética. Contudo as alterações analíticas encontradas colocam-na num estadio R-ISS de II/III correspondendo a uma sobrevida a 5 anos de 62%-40% 10.
Apesar de descrito como um subtipo de MM com mau prognóstico sabe-se atualmente que o MM IgD tem prognóstico semelhante ao dos outros subtipos quando utilizada quimioterapia à base de bortezomib e autotransplante mesmo na presença de doença extramedular 4,5. Apesar disso, o número de recaídas parece ser superior neste subtipo e a idade avançada ao diagnóstico (>65 anos), presença de alterações genéticas, plasmocitomas extramedulares e valores de β2-microglobulina elevados parecem ser fatores de mau prognóstico 5,8. A doente deste caso clínico obteve remissão completa ao 4º ciclo de quimioterapia, no entanto apresenta prognóstico reservado dado a idade avançada, a presença de múltiplos plasmocitomas extramedulares e elevados níveis de β2-microglobulina.
Em conclusão MM IgD é um subtipo raro de MM com características clínicas diferentes dos restantes subtipos, grande agressividade e associado a pior prognóstico, mas só por si não parece ser um parâmetro independente de mau prognóstico7,8.
Figura I
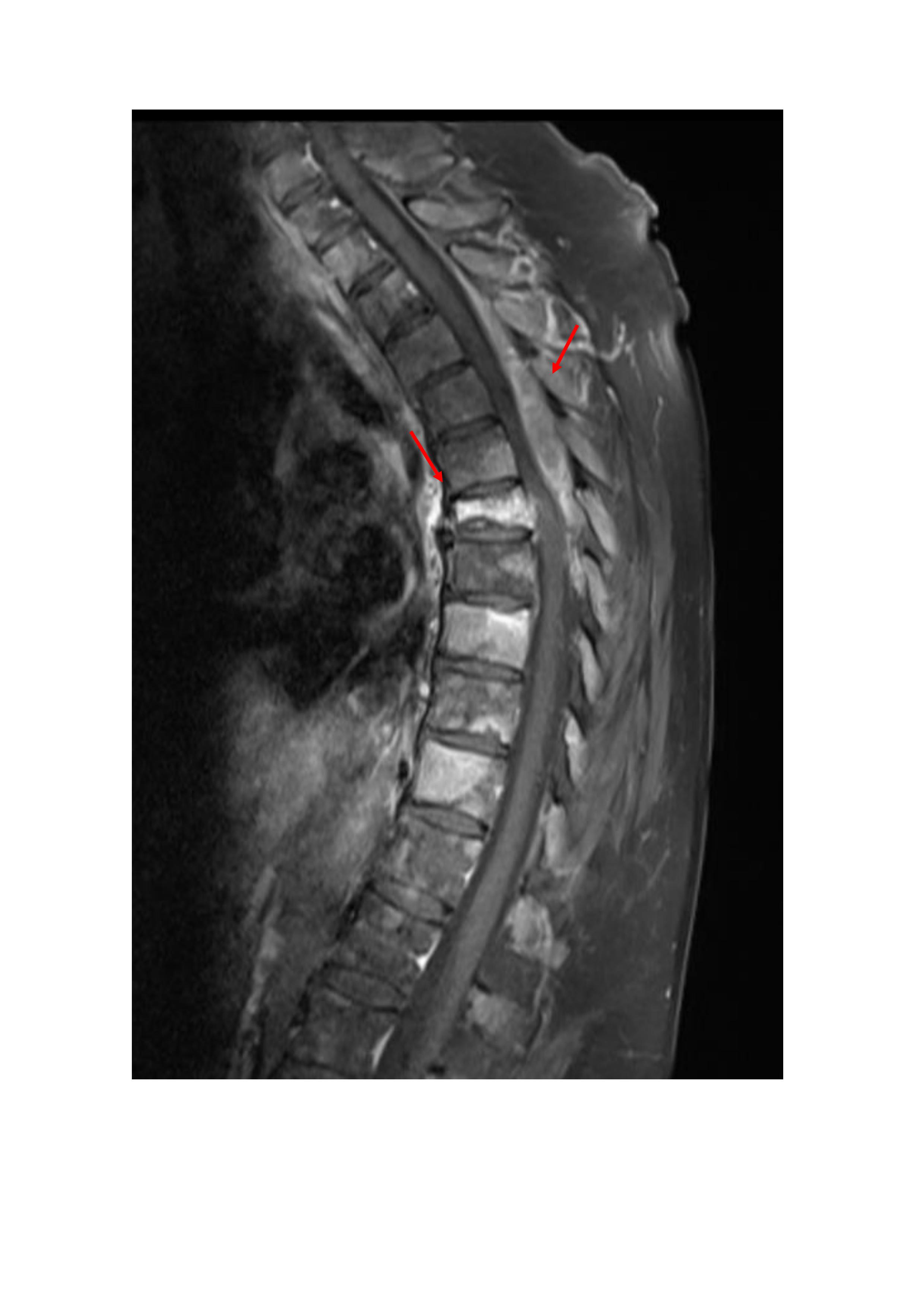
M da coluna (T1 contraste) com invasão de vários corpos vertebrais, fratura patológica de D6 e massa epidural compressiva.
Figura II
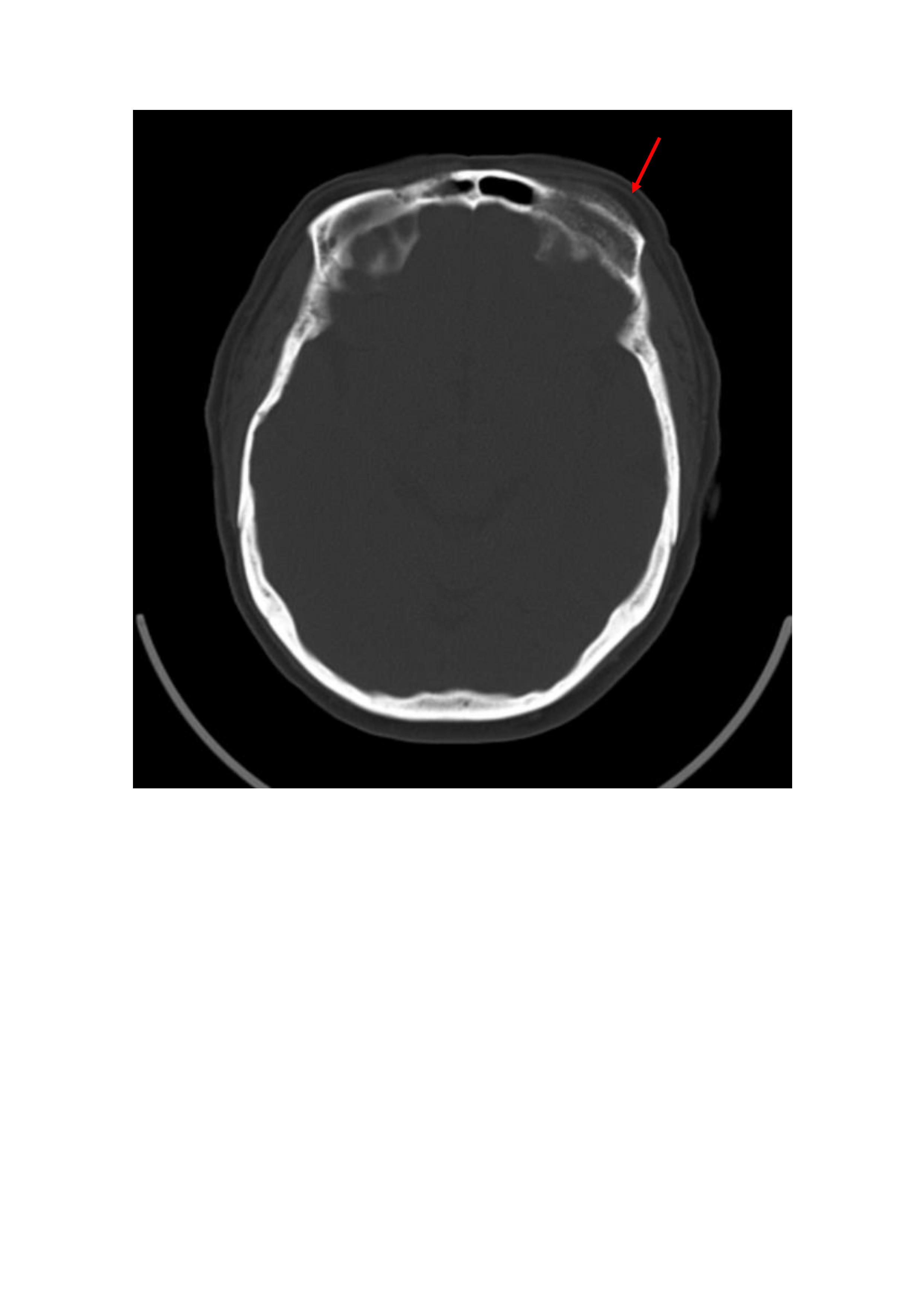
TC-CE com lesão expansiva na calote frontal anterior esquerda.
Figura III
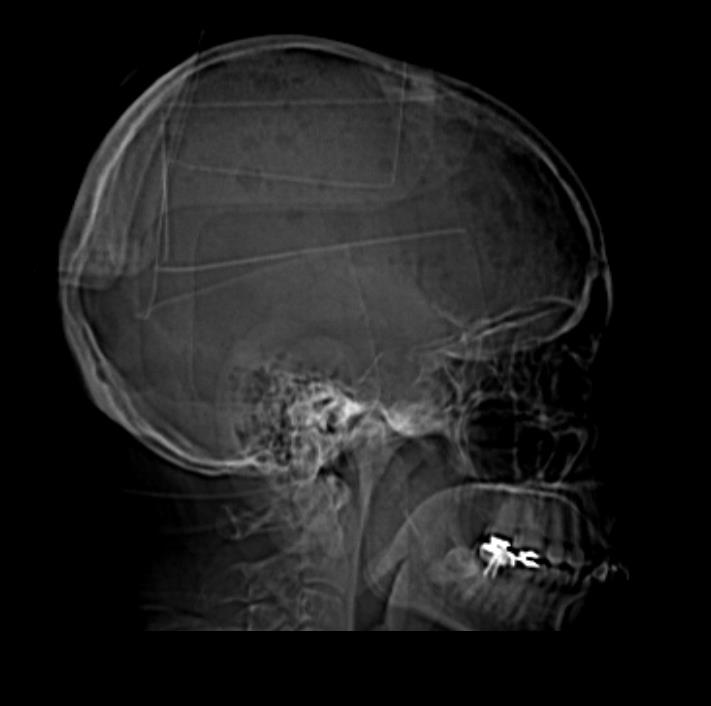
Múltiplas lesões líticas da calote craniana.
BIBLIOGRAFIA
1. Djidjik R, Lounici Y, Chergeulaïne K, Berkouk Y, Mouhoub S, Chaib S, et al. IgD multiple myeloma: Clinical, biological features and prognostic value of the serum free light chain assay. Pathol Biol. 2015;63(45):2104.
2. Gerecke C, Fuhrmann S, Strifler S, Schmidt-Hieber M, Einsele H, Knop S. The Diagnosis and Treatment of Multiple Myeloma. Dtsch Aerzteblatt Online. 2016;113:4707.
3. Zagouri F, Kastritis E, Symeonidis AS, Giannakoulas N, Katodritou E, Delimpasi S, et al. Immunoglobulin D myeloma: Clinical features and outcome in the era of novel agents. Eur J Haematol. 2014;92(4):30812.
4. Maisnar V, Hájek R, čudla V, Gregora E, Büchler T, Tichý M, et al. High-dose chemotherapy followed by autologous stem cell transplantation changes prognosis of IgD multiple myeloma. Bone Marrow Transplant. 2008;41(1):514.
5. Bladé J, Fernández De Larrea C, Rosiñol L, Cibeira MT, Jiménez R, Powles R. Soft-tissue plasmacytomas in multiple myeloma: Incidence, mechanisms of extramedullary spread, and treatment approach. J Clin Oncol. 2011;29(28):380512.
6. Shivlal Pandey, MBBSRobert A. Kyle M. Unusual Myelomas: A Review of IgD and IgE Variants [Internet]. 2013. Available from: http://www.cancernetwork.com/hematologic-malignancies/unusual-myelomas-review-igd-and-ige-variants
7. Wang GR, Sun WJ, Chen WM, Huang ZX, Zhang JJ, An N, et al. Immunoglobulin D Multiple Myeloma: Disease Profile, Therapeutic Response, and Survival. Acta Haematol. 2016;136(3):1406.
8. Kim MK, Suh C, Lee DH, Min CK, Kim SJ, Kim K, et al. Immunoglobulin D multiple myeloma: Response to therapy, survival, and prognostic factors in 75 patients. Ann Oncol. 2011;22(2):4116.
9. Moreau P, San Miguel J, Sonneveld P, Mateos M V., Zamagni E, Avet-Loiseau H, et al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(March):iv52-iv61.
10. Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, et al. Revised international staging system for multiple myeloma: A report from international myeloma working group. J Clin Oncol. 2015;33(26):28639.

