Introdução
A hemofilia adquirida é uma doença que se caracteriza pela existência de inibidores adquiridos da coagulação (autoanticorpos que inibem a atividade de um fator ou promovem a sua eliminação).1 Manifesta-se clinicamente através de diátese hemorrágica grave, normalmente espontânea. No entanto, o padrão de hemorragia é diferente do padrão clássico da hemofilia congénita (hemartroses, hematomas musculares profundos), predominando equimoses, hematomas ou hemorragia retroperitoneal. Os autoanticorpos mais comuns que afectam a cascata da coagulação, e estão associados a diátese hemorrágica são dirigidos contra o fator VIII, denominando-se por hemofilia A adquirida. A fisiopatologia da doença não está totalmente esclarecida, mas envolve polimorfismos genéticos e uma população de linfócitos T CD4+ autoreactivos.2
A incidência da hemofilia adquirida está estimada em 1.3 a 1.5 casos por milhão/ano, sendo mais comum após a sexta década de vida (com exceção dos casos associados a gravidez e puerpério).3,4 Outras causas identificáveis são lúpus eritematoso sistémico, doença oncológica e reações medicamentosas (cada responsável por 5 a 10% dos casos). Contudo em 50% dos doentes, não se identifica patologia subjacente.2
Considerando a raridade e gravidade da patologia, foram elaboradas normas de orientação clínica para o diagnóstico diferencial de hemofilia adquirida na presença de um quadro hemorrágico agudo, acompanhado por um prolongamento inexplicado do Tempo de Tromboplastina Parcial Activada (APTT).1, 5
O desafio diagnóstico aumenta quando as manifestações clínicas e as alterações analíticas da hemofilia A adquirida surgem num doente hipocoagulado com varfarina. O quadro hemorrágico pode ser atribuído ao agente dicumarínico, tornando o diagnóstico da hemofilia mais moroso. Casos semelhantes já se encontram descritos na literatura.69 Apresenta-se um caso de uma doente com quadro hemorrágico espontâneo, secundário a hemofilia A adquirida, inicialmente atribuído a intoxicação dicumarínica.
Caso clínico
Mulher de 65 anos, leucodérmica, reformada, recorreu ao Serviço de Urgência por queixas de dor na região inguinal esquerda, equimoses e tumefações em ambos os membros inferiores, de aparecimento súbito, que condicionavam limitação da mobilidade. Sem história de trauma. Trata-se de uma doente com antecedentes de acidente vascular isquémico de etiologia cardioembólica, fibrilhação auricular permanente e insuficiência venosa crónica. Hipocoagulada com varfarina há cerca de 9 anos, sem complicações hemorrágicas major. Estava medicada com perindopril, furosemida e amiodarona.
Ao exame objectivo apresentava-se normotensa, com ritmo irregular e frequência cardíaca de 92 batimentos por minuto. Abdómen mole, depressível, indolor, sem massas e com pontos herniários livres. Membros inferiores com equimoses dispersas, tumefação do terço proximal da face interna da coxa esquerda, não pulsátil e sem sinais inflamatórios. Do estudo analítico realizado (Tabela 1) evidencia-se anemia (Hemoglobina 6.0g/dL) normocítica e normocrómica, plaquetas sem alterações quantitativas, aumento do tempo de protrombina, do INR (TP= 56.9 segundos, INR 4.6) e Aptt (81.3 segundos).
Foi submetida TC abdominal e pélvica (Fig. 1) que revelou
densificação retroperitoneal para-renal esquerda, associada a aumento da espessura do músculo psoas ilíaco ipsilateral, com densitometria heterogénea e áreas hipodensas que perfazem 19X15 cm no plano sagital. Considerando a hipocoagulação terapêutica, a alterações admitem-se estar em relação com alterações hemorrágicas espontâneas. No plano muscular da porção proximal da coxa esquerda, destacam-se colecções hipodensas de 8,1cm de diâmetro total, em relação com a mesma etiologia
Optou-se por terapêutica conservadora, com a suspensão da varfarina, administração de 10 mg de fitomenadiona endovenosa, e suporte transfusional com 3 unidades de plasma fresco congelado (PFC) e 2 unidades de concentrado eritrocitário (CE). Foi decidido internamento por hematoma retroperitoneal secundário a intoxicação dicumarínica.
Nas 24 horas seguintes, a doente manteve estabilidade hemodinâmica com normalização do INR e diminuição parcial dos valores APTT, destaca-se um baixo rendimento transfusional pelo que foi submetida a nova transfusão de CE ao 3º dia de internamento.
Ao 4º dia de internamento observou-se episódio súbito de instabilidade hemodinâmica com taquicardia e oligúria. O estudo analítico revelou agravamento da anemia e prolongamento mantido do APTT (43,2 segundos). Perante suspeita clínica de deficit de fator da coagulação, iniciou PFC fixo (10mL/kg,) e foi submetida a transfusão de 2 unidades CE. Apesar de não constituir terapêutica de 1ª linha, optou-se pelo hemoderivado supracitado devido à necessidade de tratamento urgente e à ausência em tempo útil de complexo protrombínico. Verificou-se estabilidade hemodinâmica, obtendo-se a confirmação do deficit de fator VIII (fator VIII 11, valores de referência 50-150) e a presença de um inibidor (8 unidades de Bethesda - UB) (Fig. 2), pelo que iniciou terapêutica dirigida ao inibidor com prednisolona (1mg/kg). Ao 13ª dia de internamento destaca-se quadro súbito de taquicardia, hipertensão e sinais de sobrecarga hídrica, com resposta à terapêutica médica e ventilação mecânica não-invasiva que se admitiram como provável insuficiência cardíaca de predomínio esquerdo.
Devido ao alto risco hemorrágico e de disfunção multiorgânica optou-se pela transferência hospitalar. Na unidade hospitalar terciária realizou 1 dose de complexo protombínico ativado (FEIBA® 50 unidades/kg peso), com a ausência de novos episódios de hemorragia, recuperação paulatina dos valores de hemoglobina, diminuição do APTT e aumento dos níveis de fator VIII. Destaca-se inibidor negativo ao 5º dia de corticoterapia.
Discussão
A hemofilia A adquirida é uma doença hemorrágica rara, causada pela presença de um autoanticorpo que neutraliza a ação hemostática do fator VIII. A maioria dos casos é idiopática, mas pode estar associada à gravidez (entre o primeiro e quarto més do pós-parto), a doença hemato-oncológica (leucemia linfocítica crónica, mieloma múltiplo, linfoma não Hodgkin), a neoplasias de órgão sólido (pulmão, próstata, pancreas), doenças autoimunes (lúpus eritematoso sistémico, artrite reumatóide, síndrome de Sjogren) e a fármacos (antibióticos, fenitoína, metildopa). Atribui-se a origem dos auto-anticorpos à perda dos mecanismos de tolerância imunitária, que regulam a resposta endógena ao fator VIII. A existência de células T CD4+ específicas para fator VIII têm um papel central na fisiopatologia da doença.
Laboratorialmente verifica-se o aumento isolado do APTT, que não é corrigido através da mistura com plasma normal (teste de mistura), com o INR normal. Além de deficits de fatores de coagulação (que são excluídos por correção do APTT após o teste de mistura), importa excluir a presença de anticoagulante lúpico (que normalmente cursa com eventos trombótico) e uso de heparina.10 O diagnóstico de hemofilia A adquirida é confirmado pela documentação da diminuição da actividade do fator VIII e pela presença de um inibidor do respectivo fator, quantificado pelo ensaio de Bethesda (Fig. 3). Neste teste, a actividade do inibidor é medida através de diluições progressivas do plasma com inbidor, em plasma sem alterações. Os doentes com hemofilia adquirida são classificados como tendo um título baixo (< 5 UB) ou título alto de inibidor (>5 UB).
Um quadro de hemorragia aguda em doentes sob varfarina e INR supraterapêutico é facilmente atribuível à ação do agente dicumarínico. A varfarina aumenta o TP e o APTT, não existindo valores de APTT de referência para doentes sob tratamento com varfarina.11 Um aumento desproporcional do APTT em relação ao INR (especialmente se entre 2-3) pode constituir um indício de outra coagulopatia subjacente. Por sua vez, a prevalência da hemofilia adquirida bem como das patologias com indicações para a hipocoagulação aumenta com a idade, com maior probabilidade da hemofilia se encontrar mascarada pela varfarina.
O tratamento da hemofilia adquirida engloba 3 objetivos complementares: o controlo e prevenção da hemorragia, a erradicação do inibidor e o tratamento da patologia causal (se existir).1 Os doentes com hemorragia major ou diminuição dos valores de hemoglobina necessitam de tratamento hemostático. Os agentes hemostáticos de primeira linha incluem fator VIII suíno recombinante (terapêutica de substituição), concentrado de complexo protrombínico ativado ou Fator VII recombinante ativado (ambos terapêutica de ponte).2,12 O uso de desmopressina deve ser reservado para doente com hemorragia ligeira. O fator VIII humano recombinante é eficaz apenas no tratamento da hemofilia adquirida em doentes com titulo baixo de inibidor (< 5 UB). A erradicação do inibidor (auto anticorpos contra o fator VII) é alcançada através da imunossupressão através de corticoides isoladamente, ou em associação com ciclofosfamida ou rituximab, durante pelo menos 3 semanas (Fig. 4).1,12
No caso apresentado, um hematoma retroperitoneal espontâneo, foi inicialmente atribuído a uma intoxicação dicumarínica. Apesar da elevação persistente do APTT, a hipótese da sua relação com a varfarina levou a um atraso no diagnóstico, a um tratamento inapropriado e à referenciação tardia para a Hematologia.
Num doente com hemorragia associada a sobredosagem de varfarina, deve ser pedido tanto o TP como o APTT. Critérios clínicos orientam as medidas terapêuticas, mas a resposta a estas intervenções deve ser monitorizada.
Apresentações prévias
Comunicação oral no 24º Congresso Nacional de Medicina Interna.
Quadro I
Exames complementares de diagnóstico realizados no serviço de urgência.
| | | |
| Estudo analítico | Resultado | Valor de Referencia |
| Hemoglobina | 6.0 g/dL | 12.0-15.0 |
| VGM | 97.5 fL | 83.0-101.0 |
| Leucócitos | 9.6 X 10E9/L | 4.0-10.0 |
| Plaquetas | 273 X 10E9/L | 150-400 |
| Tempo protrombina | 56.9 segundos | 10.0-13.0 |
| INR | 4.6 | 0.88-1.12 |
| aPTT | 81.3 segundos | 25.0-35.0 |
| Ureia | 120 mg/dL | 21.0-43.0 |
| Creatinina | 2.0 mg/dL | 0.6-1.1 |
| LDH | 343 U/L | 125-220 |
| Bilirrubina | 0,9mg/dL | 0,2-1.2 |
| PCR | 3.97 mg/dL | < 0.50 |
Figura I
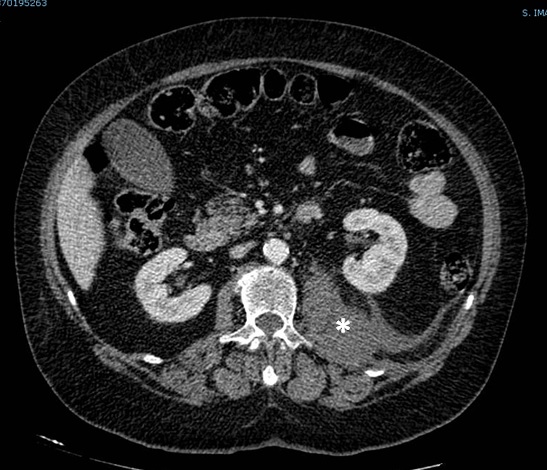
AngioTC abdominal e pélvica - densificação retroperitoneal para-renal esquerda (*)
Figura II
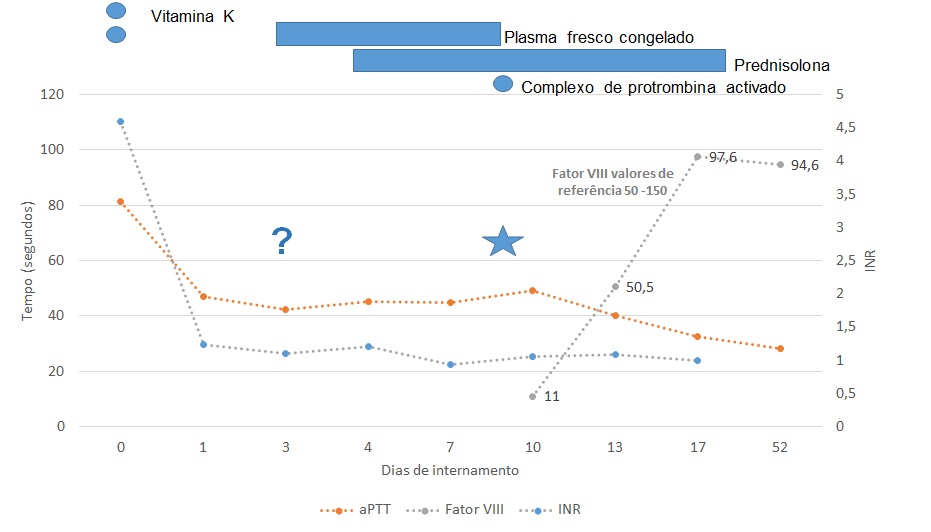
Evolução analítica (APTT, INR e fator VIII), ao longo do internamento. Representação temporal da terapêutica. (?) suspeita de deficit de fator de coagulação; (Estrela) confirmação da existência de inibidor.
Figura III
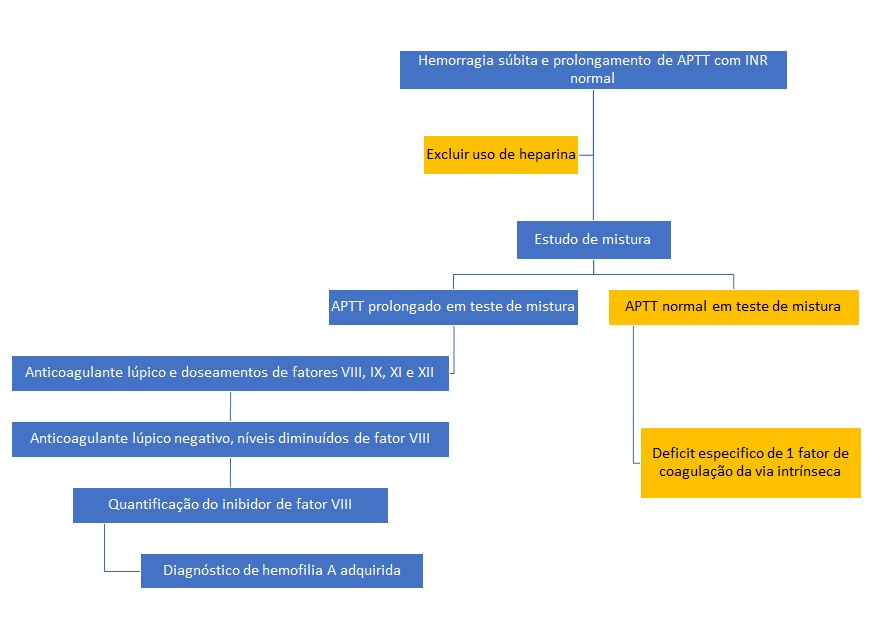
Algoritmo diagnóstico da hemofilia A aquirida.1
Figura IV
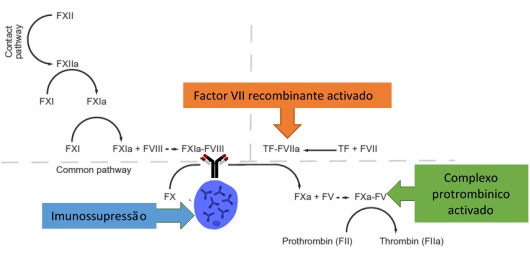
Opções terapêuticas usadas na hemofilia adquirida. Os agentes de ponte (Factor VII recombinante e complexo protrombínico activados) activam a cascata de coagulação a jusante do inibidor. A imunossupressão impede a sua produção.
BIBLIOGRAFIA
1. Kruse-Jarres R, Kempton CL, Baudo F, Collins PW, Knoebl P, Leissinger CA, et al. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am J Hematol. 2017 Jul;92(7):695705.
2. Franchini M, Vaglio S, Marano G, Mengoli C, Gentili S, Pupella S, et al. Acquired hemophilia A: a review of recent data and new therapeutic options. Hematology. 2017 Oct 21;22(9):51420.
3. Collins P, Macartney N, Davies R, Lees S, Giddings J, Majer R. A population based, unselected, consecutive cohort of patie with acquired haemophilia A. Br J Haematol. 2004;
4. Knoebl P, Marco P, Baudo F, Collins P, Huth-Kühne A, Nemes L, et al. Demographic and clinical data in acquired hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012;
5. W Collins P, Chalmers E, Hart D, Jennings I, Liesner R, Rangarajan S, et al. Diagnosis and management of acquired coagulation inhibitors: A guideline from UKHCDO. Br J Haematol. 2013;
6. Uggla B, Linder O, Schulman S. Acquired hemophilia masked by warfarin therapy: report on two cases. Blood Coagul & fibrinolysis an Int J Haemost Thromb. 2003;
7. Kantor R, Mayan H, Puritz L, Varon D, Farfel Z. Acquired hemophilia masked by warfarin therapy. Am J Med Sci. 2000;
8. Vadikolia CM, Riddell A, Brooks S, Yee TT, Brown S, Lee C. Acquired haemophilia masked by warfarin therapy. Int J Lab Hematol. 2007;
9. Lawless S, Benson G. A 75-year-old woman with acquired haemophilia disguised by warfarin treatment. BMJ Case Rep. 2015;
10. Tiede A, Werwitzke S, Scharf R. Laboratory Diagnosis of Acquired Hemophilia A: Limitations, Consequences, and Challenges. Semin Thromb Hemost]. 2014 Oct 9;40(07):80311.
11. Hauser VM, Rozek SL. Effect of warfarin on the activated partial thromboplastin time. Drug Intell Clin Pharm. 1986;
12. Ma AD, Kessler CM, Al-Mondhiry HAB, Gut RZ, Cooper DL. Use of recombinant activated factor VII for acute bleeding episodes in acquired hemophilia. Blood Coagul Fibrinolysis. 2016 Oc;27(7):75360

