Introdução
A macroglobulinemia de Waldenström (MW) é uma doença linfoproliferativa rara que se carateriza pela produção de imunoglobulina M (IgM) pentamérica.1 Representa 1 a 2% de todos os linfomas não-Hodgkin, afetando maioritariamente homens idosos, com uma mediana de idade que varia entre 63 e 75 anos.2-4
A apresentação clínica da MW é extremamente heterogénea, o que torna o diagnóstico desafiante. Os sintomas podem ser atribuídos à infiltração pelas células clonais (anemia, linfadenopatia e esplenomegalia) ou causados pela IgM monoclonal (neuropatia, hiperviscosidade e crioglobulinemia), sendo que um número substancial de doentes se encontra assintomático no momento do diagnóstico.5
Caso clínico
Apresentamos um homem de 71 anos com antecedentes de colectomia total, em 2008, por adenocarcinoma do cólon moderadamente diferenciado, T3N0Mx, sem evidência de recidiva. Recorreu ao Serviço de Urgência (SU) em maio de 2018, por um quadro de hematoquézias, sem perdas hemáticas prévias, sem febre e sem perda ponderal nos últimos tempos. Ao exame físico, apresentava mucosas pálidas e encontrava-se apirético e hemodinamicamente estável. Não eram palpáveis adenopatias periféricas e o exame abdominal não apresentava alterações. No estudo analítico observava-se anemia macrocítica de novo (hemoglobina de 8.6 g/dL com VGM 101.1 fL). Assim, como a apresentação inicial foi um quadro de hemorragia digestiva, associado aos antecedentes do doente, foi admitido no Serviço de Cirurgia Geral para compensação e estudo etiológico. Durante o internamento manteve necessidade de suporte transfusional frequente para manter valores de hemoglobina entre 7.1g/dL a 8.6g/dL. Realizou endoscopia digestiva alta que mostrou angiectasias de 15 e 5 mm na grande curvatura do estômago, muito friáveis, com hemorragia ao toque; foi efetuada termoablação com árgon plasma. Realizou também coloscopia total com apenas ectasia venosa do reto. Ao 5º dia, após terapêutica endoscópica, reinicia retorragias, tendo repetido estudos endoscópicos que desta vez não evidenciaram hemorragia ativa. Foi, então, assumido hematoquézias em contexto de hemorróidas, pelo que foi realizada hemorroidectomia, e não se progrediu no restante estudo de causas de anemia. No restante tempo de internamento, manteve-se sem perdas hemáticas, pelo que teve alta orientado para consulta de Cirurgia geral.
Em julho, no contexto de dorsalgia incapacitante, foi internado no Serviço de Medicina. À admissão mantinha a anemia macrocítica (Quadro 1), sem outras alterações no estudo analítico, incluindo função renal e perfil hepático.
Completando o estudo etiológico da anemia, durante o internamento no Serviço de Medicina, efetuou eletroforese de proteínas sérica que evidenciou um pico monoclonal gamma (52%). A imunoeletroforese confirmou gamapatia monoclonal IgM/kappa, com IgM 6571mg/dL. Realizou, também, tomografia computorizada toraco-abdomino-pélvica que revelou múltiplas adenomegalias localizadas a nível para-vertebral, retrocurais, vertebrais dorsais e para aórticas, com cerca de 17 mm de maior diâmetro, assim como estruturas ganglionares milimétricas jugulocarotídeas e supra-clavículares bilaterais.
Perante estes achados, e na suspeita de síndrome linfoproliferativo, foi realizada biópsia da medula óssea (MO), cujo exame histológico mostrou medula óssea extremamente hipercelular (Figura 1), com representação das três linhas celulares hematopoiéticas e infiltrado difuso de células linfóides pequenas, plasmócitos e linfoplasmócitos (Figura 2).
O estudo imunohistoquímico mostrou positividade para CD20 (Figura 3) e Bcl2, sem reatividade para CD3, CD5, CD10, CD23, Bcl6 e ciclina D1, com as células neoplásicas a mostrar reatividade citoplasmática para cadeias leves kappa. Estes achados histológicos e imunohistoquímicos são sugestivos de linfoma não-Hodgkin de células B, linfoplasmocítico, o que, aliado ao pico IgM, estabelece o diagnóstico de MW.
Perante estes achados, doseou-se a beta 2 microglobulina pela sua importância prognóstica, sendo que um valor de 5,81 mg/L, em conjunto com a idade > 65 anos e níveis de hemoglobina ≤11,5 g/dL, colocou este doente no grupo de alto risco.
Apesar de não apresentar sintomatologia ocular, atendendo a que apresentava elevada concentração de IgM circulante, foi pedida avaliação por Oftalmologia para rastreio de alterações compatíveis com hiperviscosidade. A avaliação do fundo do olho mostrou múltiplas hemorragias intrarretinianas dispersas com ingurgitamento venoso ligeiro, não demonstrando de forma inequívoca a presença de hiperviscosidade. Neste sentido, e para descartar envolvimento cerebral, realizou tomografia cerebral que não evidenciou lesões agudas. Durante o internamento, nunca apresentou sintomas para além da dor óssea focalizada na coluna dorsal, pelo que se considerou não beneficiar de plasmaférese urgente. Foi então encaminhado para consulta de Hematologia, onde iniciou terapêutica com Rituximab, Bortezomib e Dexametasona, e de Medicina Interna. Desde então, manteve-se sempre sem sintomas de hiperviscosidade, com descida sustentada de IgM e de VS, ao fim de 4 ciclos de tratamento, com hemoglobina de 11.2g/dL, VS de 78 mm/1H e IgM de 2107.0 mg/dL.
Discussão
O sintoma de apresentação na MW mais comum é a fadiga relacionada com a anemia.6 No entanto, no presente caso clínico, esta sintomatologia foi inicialmente filiada em contexto de hemorragia digestiva, o que dificultou o diagnóstico.
Nos doentes com MW, a hipergamaglobulinemia IgM circulante parece interagir com os fatores de coagulação e revestir as plaquetas, o que pode contribuir para um aumento do risco hemorrágico.7 Esta alteração pode, então, explicar as múltiplas hemorragias digestivas como forma de apresentação da MW, num doente com angiectasias, e que recorreram, mesmo após terapêutica com árgon plasma.
Da mesma forma, a circulação de IgM monoclonal é responsável, também, pela síndrome de hiperviscosidade, que é uma manifestação comum da MW, afetando 10 a 15% dos doentes. Os sintomas mais frequentemente associados a esta síndrome são epistaxes recorrentes, cefaleias, visão turva e diminuição da acuidade visual, acufenos, vertigens e alterações do estado mental.8 Estes raramente ocorrem em doentes com níveis de IgM inferiores a 3000 mg/dL. A medição da viscosidade sérica pode ser considerada em doentes com estes sintomas, mas não se correlaciona com a gravidade desta síndrome. O exame do fundo do olho a mostrar o ingurgitamento das veias da retina é um sinal mais confiável de hiperviscosidade clinicamente relevante. A plasmaférese deve ser prontamente iniciada em doentes com sinais e sintomas de hiperviscosidade, no entanto, é usada de forma temporária, até que a terapêutica definitiva tenha sido instituída.9 Outros sintomas de apresentação relacionados à IgM monoclonal incluem crioglobulinemia, anemia hemolítica (~5% dos doentes), neuropatia periférica (em 5-10%) e amiloidose (em 2%).7
O diagnóstico de MW baseia-se na confirmação histológica da infiltração da MO por elementos linfoplasmocíticas e a deteção de gamapatia monoclonal IgM sérica.11,12 A população clonal de células linfoplasmocíticas presentes na MO tipicamente expressa CD19, CD20, CD22 e CD79a, o que pode auxiliar o diagnóstico. 13,14 No presente caso clínico, os linfócitos presentes na MO expressavam CD20 e Bcl2, o que, em conjunto com os outros achados, permitiu firmar o diagnóstico de MW.
Na avaliação de um doente com MW, é importante a avaliação do hemograma completo assim como o doseamento da beta 2 microglobulina, que funcionam como marcadores de prognóstico, permitindo a classificação dos doentes em diferentes níveis de risco (baixo, intermédio, alto), correspondendo a taxas de sobrevivência aos 5 anos de, respetivamente, 87%, 68% e 36%. 15 O doente aqui apresentado foi incluído no nível de risco alto.
O tratamento é reservado aos que se encontram sintomáticos, sendo a anemia a razão mais comum para iniciar tratamento. Esta pode ser causada por invasão da medula óssea, défice de ferro e/ou hemólise.16 Neste caso, foram descartadas as últimas duas hipóteses, restando como causa da anemia a invasão da MO.
Conclusão
A MW é uma patologia rara e de diagnóstico complexo. O presente caso clínico pretende demonstrar a complexidade do diagnóstico de MW num doente com diversos fatores confundidores, demonstrando a importância de investigar a etiologia da anemia refratária.
Quadro I
Estudo analítico à admissão (julho)
| | | | | | |
| | | | | | |
| Eritrograma | | Valores de referência | Química Clínica | | Valores de referência |
| Hemoglobina g/dL | 7,1 | 13-18 | Ferro µL/dL | 42 | 31-144 |
| Eritrócitos Exp6/µL | 2,19 | 4,4 -5,9 | Capacidade de fixação µL/dL | 210 | 228-428 |
| Hematócrito % | 23 | 40 - 52 | Saturação de transferrina % | 20 | 20-55 |
| VGM fL | 105 | 80-100 | Ferritina ng/mL | 138,60 | 21,81-274,66 |
| HGM pg | 32,4 | 26-34 | Vitamina B12 pg/mL | 655 | 189-883 |
| CHGM g/dL | 30,9 | 32-36 | Folatos ng/mL | 7,90 | 2,34-18,99 |
| RDW % | 17,7 | 11,5-14,5 | | | |
| Esfregaço de sangue periférico: sem alterações nas três linhagens | | | Hemoterapia | | |
| | | | Coombs direto | Negativo | |
| | | | Coombs indireto | Negativo | |
| | | | INR | 1.3 | |
| | | | Tempo de Cefalina (aPTT) seg | 35,1 | Normal do dia 27,7 |
| | | | Tempo de Tombina seg | 12,6 | 11,8-17,6 |
Figura I
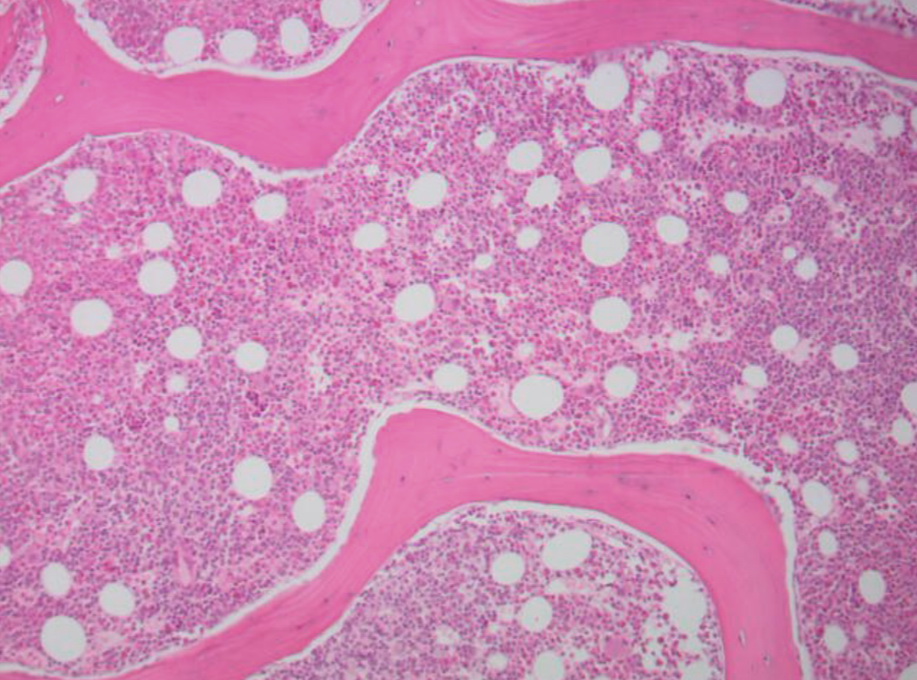
Medula óssea hipercelular
Figura II
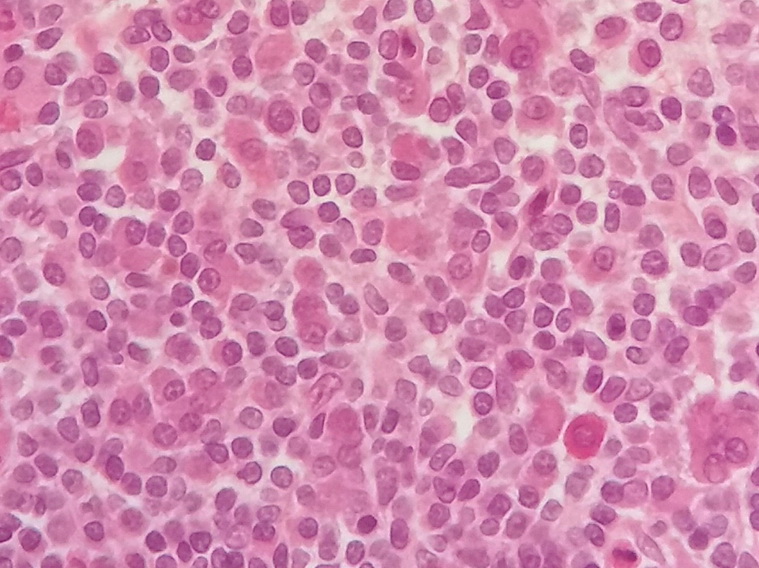
Infiltrado difuso de células linfóides pequenas, plasmócitos e linfoplasmócitos
Figura III
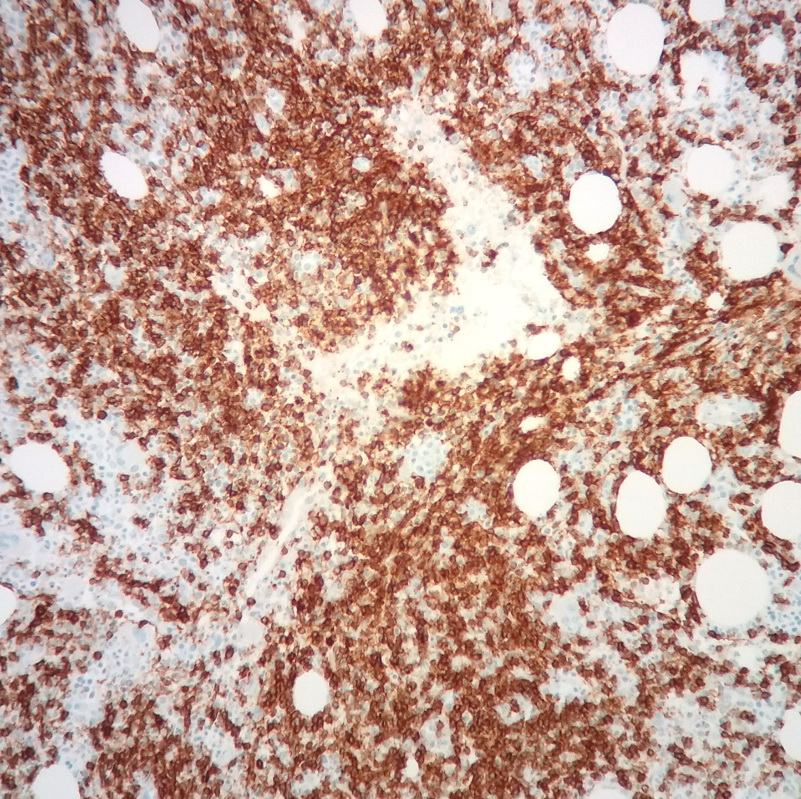
Estudo imunohistoquímico positivo para CD20
BIBLIOGRAFIA
1. Waldenström J. Incipient myelomatosis or essential hyperglobulinemia with fibrinogenopenia: a new syndrome? Acta Med Scand 1944; 117:216- 247.
2. Herrinton L, Weiss N. Incidence of Waldenström macroglobulinemia. Blood 1993; 82: 3148-3150.
3. Teras L, DeSantis C, Cerhan J, Morton L, Jemal A, Flowers C. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016; 66: 443459.
4. Treon S. How I treat Waldenström macroglobulinemia. Blood 2009; 114: 23752385.
5. Dimopoulos M, Kyle R, Anagnostopoulos A, Treon S. Diagnosis and management of Waldenström macroglobulinemia. J Clin Oncol 2005; 23:1564-77.
6. Björkholm M, Johansson E, Papamichael D, Celsing F, Matthews J, Lister TA, et al. Patterns of clinical presentation, treatment, and outcome in patients with Waldenströms macroglobulinemia: A two-institution study. Semin Oncol 2003;30:226230.
7. Vijay A, Gertz M. Waldenström macroglobulinemia. Blood 2007; 109: 5096-5103.
8. Stone M, Bogen S. Evidence-based focused review of management of hyperviscosity syndrome. Blood 2012; 119:2205-8.
9. Menke M, Feke G, McMeel J, Treon S. Ophthalmologic techniques to assess the severity of hyperviscosity syndrome and the effect of plasmapheresis in patients with Waldenström macroglobulinemia. Clin Lymphoma Myeloma 2009; 9: 100103.
10. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Vol. 2 (Ed Revised 4th Edition). Geneva: WHO Press 2017.
11. Kyle R, Treon S, Alexanian R, Barlogie B, Björkholm M, Dhodapkar M, et al. Prognostic markers and criteria to initiate therapy in Waldenström macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenström macroglobulinemia. Semin Oncol 2003; 30: 116120.
12. McMaster M. Familial Waldenström macroglobulinemia. Semin Oncol. 2003;30:146-152.
13. Treon S, Hunter Z, Aggarwal A, Ewen E, Masota S, Lee C, et al. Characterization of familial Waldenström macroglobulinemia. Ann Oncol. 2006;17:488-494.
14. Morel P, Duhamel A, Gobbi P, Dimopoulos M, Dhodapkar M, McCoy J, et al. International prognostic scoring system for Waldenström macroglobulinemia. Blood 2009; 113: 4163-7.
15. Kastritis E, Leblond V, Dimopoulos M, Kimby E, Staber P, Kersten M, Tedeschi A, et al. Waldenströms macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2018; 0: iv1iv10.

