

INTRODUÇÃO
As Microangiopatias trombóticas (MAT) constituem um grupo de patologias muito diverso, com um conjunto de características clínicas comuns: anemia hemolítica microangiopática (AHM); trombocitopenia e lesão de órgão (por trombose microvascular, através de trombos ricos em plaquetas e do espessamento das paredes das arteríolas/capilares).1
Classicamente, as MAT primárias dividiam-se em Púrpura trombocitopénica trombótica (PTT) - Congénita/adquirida; e Síndrome hemolítico-urémico (SHU) - Típico/atípico. 1,3,4,5 No entanto, em 2014, foi sugerida por George J. uma nova classificação, que inclui nove distúrbios (Tabela 1).5 Estas podem surgir após a exposição a um factor precipitante (infecções, pancreatite, HIV, gravidez/pós-parto, vacinação, fármacos, álcool), em doentes com predisposição.1,3,5,7 As MAT secundárias (Tabela 2), cujos sinais/sintomas podem inicialmente sugerir o diagnóstico de MAT primária, devem ser sempre excluídas, uma vez que a resolução das mesmas passa pelo tratamento dirigido à patologia de base.3,4,5
Este artigo refere-se a um caso clínico de Púrpura Trombocitopénica Trombótica.
A PTT é uma entidade rara, resultante do défice da actividade da Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13 (ADAMTS13), adquirido (95%) ou hereditário (5%). A ADAMTS13 consiste numa protease clivadora do factor von Willebrand (fvW) libertado pelas células endoteliais. Na ausência desta proteína, permanecem no plasma grandes múltimeros de fvW, com actividade trombogénica. O diagnóstico de PTT requer a documentação de défice de actividade da ADAMTS13. A PTT adquirida é um distúrbio auto-imune causado por um anticorpo inibidor da actividade da ADAMTS13, ao contrário da PTT hereditária, em que o anticorpo está ausente. O diagnóstico de PTT hereditária/Síndrome Upshaw-Schulman implica a documentação de uma mutação do gene ADAMTS13.1,3,5,7
CASO CLÍNICO
Apresentamos um caso de uma jovem de 23 anos, caucasiana. Previamente saudável, cuja única medicação habitual incluía um contraceptivo oral. Descrevia uma eventual história familiar de doenças hematológicas (primo paterno em segundo grau falecido na infância com púrpura (sic) e tia paterna com episódio de trombocitopenia na gravidez).
A doente apresentava queixas com uma semana de evolução de astenia em agravamento, dispneia para pequenos esforços, palpitações, menorragia abundante, noção de palidez e escleróticas ictéricas. Referia ainda um episódio de 3 dejecções líquidas durante um dia e odinofagia, aproximadamente três semanas antes do início do quadro actual.
Ao exame objectivo encontrava-se: ictérica e pálida; subfebril; taquicárdica mas normotensa; com petéquias nos membros inferiores e algumas equimoses dispersas. O restante exame físico não apresentava alterações de relevo.
Do estudo efectuado à admissão (Tabela 3), apresentava critérios de anemia hemolítica microangiopática (Hg 6.4g/dL; BT 6.6mg/dL; LDH 2009U/L; haptoglobina <8mg/dL); trombocitopenia grave (Plaq <10000/uL); PCR (22,3 mg/L) e VS elevadas (61mm/1ªh); com função renal, exame de urina, estudo da coagulação e perfil hepático normais. Realizou ecografia abdomino-pélvica e tomografia computorizada (TC) toracoabdominopélvico que não mostraram alterações (sem organomegalias).
Após a admissão, foram identificados episódios recorrentes e transitórios de défices neurológicos focais: hipostesia no hemicorpo esquerdo e disartria. Nesse contexto foi efectuada uma TC cranioencefálica que se revelou normal.
Perante o quadro clínico e tendo em conta que a doente apresentava um PLASMIC score8 de 7 (todos os parâmetros preenchidos: trombocitopenia <30000/uL (10000/uL), critérios de hemólise (haptoglobina indoseável, bilirrubina indirecta de 5,68mg/dL e reticulócitos 8,5%), International Normalised Ratio (INR) <1,5 (1,0), creatinina <2mg/dL (0,63mg/dL), volume corpuscular médio <90 fl (80 fl) e ausência de neoplasia activa no último ano ou de história de transplante de órgão ou medula óssea), o que lhe confere um risco elevado de défice severo de ADAMTS13 (6282%), foi assumido o diagnóstico de provável Púrpura Trombocitopénica Trombótica.
Foi admitida em Unidade de Cuidados Intermédios, onde iniciou pulsos de metilprednisolona 1g/dia durante três dias, seguidos de 125mg de 12/12h e desmame subsequente com prednisolona. Iniciou também sessões de plasmafiltração com reposição de plasma fresco, tendo efectuado um total de seis sessões inicialmente diárias.
Em relação ao estudo etiológico, foi efectuado mielograma que mostrou medula reactiva, sem evidência de doença medular primária; serologias víricas, exames culturais, estudo imunológico, protrombótico e de outras causas de anemia, que revelaram apenas um ligeiro défice de vitamina B12 e folatos (Tabela 4). Efectuou também uma ressonância magnética cranioencefálica que revelou lesões isquémicas agudas na substância branca cerebral subcortical occipital e parietal à direita, confirmando o envolvimento neurológico.
Verificada evolução clínica e analítica favorável, com normalização dos valores de plaquetas e DHL, o que permitiu espaçar e posteriormente suspender as sessões de plasmafiltração (9º dia de internamento) (Figura 1). À data de transferência para a enfermaria geral, encontrava-se sem défices neurológicos; analiticamente: Hg 9.4g/dL; Plaq 181000/uL; BT 1.0mg/dL; LDH 220U/L; haptoglobina 27mg/dL PCR 0.60mg/dL. Teve alta para a consulta externa de Medicina Interna, onde se mantém estável, sem recidiva de doença.
Do estudo diagnóstico de PTT, foi constatado um défice de actividade de ADAMTS13 (15% - normal: 40-130) com ausência de anticorpo anti-ADAMTS13 (5U/mL normal: <15). No entanto, estes resultados dizem respeito a uma colheita efectuada posteriormente à resolução do quadro (fora da fase aguda e após tratamento), uma vez que a amostra colhida no dia da admissão na unidade de cuidados intermédios não foi devidamente conservada e não foi testada. Por outro lado, o estudo genético para mutação do gene ADAMTS13 foi negativo. Posto isto, não foi possível classificar devidamente o tipo de PTT manifestado nesta doente. Porém, se estes resultados (défice de actividade ADAMTS13 sem evidência de anticorpo anti-ADAMTS13 ou de mutação genética) se confirmassem antes do início de tratamento, este quadro seria classificado como PTT adquirida de mecanismo indeterminado, segundo o que descrevem Joly et al. numa revisão de 2017.7
DISCUSSÃO
A apresentação clínica das PTT é muito variável (tabela 5). Antes do surgimento de um tratamento eficaz, o diagnóstico baseava-se numa pêntade de sinais e sintomas: AHM, trombocitopenia, febre, alterações neurológicas e renais. No entanto, o reconhecimento da eficácia da plasmafiltração implicou a simplificação dos critérios de diagnóstico, para um início de tratamento mais rápido. Sabe-se que o envolvimento neurológico não acontece em todos os doentes (60%) e pode ser apenas transitório; a disfunção renal é mais típica do SHU e rara na PTT (10-27%)3,5,7 e que a febre ocorre em 23-50% dos doentes.4 Menos frequentemente, ocorre trombose microvascular cardíaca (~25%) ou gastrointestinal (~53).4,7 Posto isto, passou apenas a considerar-se como critérios de diagnóstico a presença de AHM e trombocitopenia, sem outra causa aparente.1,2,3,5
O diagnóstico requer o doseamento sérico da actividade da ADAMTS13 e do anticorpo inibidor da ADAMTS13. A colheita pré-tratamento é fundamental, uma vez que a plasmafiltração com infusão de plasma remove os anticorpos anti-ADAMTS13 e repõe os níveis de actividade da ADAMTS13. No entanto, o doseamento da actividade de ADAMTS13 não é suficientemente sensível para identificar os doentes com PTT nem suficientemente específico para excluir doentes com outros diagnósticos.
Neste caso, apesar de, do ponto de vista clínico, ser altamente compatível com um quadro de PTT, o mecanismo responsável pelo quadro não foi determinado laboratorialmente, por falha no timing das colheitas. Por outro lado, na presença de um défice de vitamina B12, faria sentido efectuar o doseamento de homocisteína e ácido metilmalónico para exclusão de MAT mediada por alterações do metabolismo (o que poderá ser assumido como limitação do artigo). No entanto, este défice era ligeiro e a doente recuperou totalmente com a terapêutica instituída dirigida ao diagnóstico de PTT, sem ter sido realizada suplementação parentérica com vitamina B12 (terapêutica preconizada para MAT por alterações do metabolismo da vitamina B12), o que torna a hipótese deste diagnóstico pouco sustentável.
É importante investigar a etiopatogenia e o tipo de MAT, mas é mais importante ainda iniciar o tratamento precocemente (<4-8h após o diagnóstico).3 Com a plasmafiltração, a sobrevivência ronda os 80-90%.1,3,5 Anteriormente, a sobrevida era de apenas 10%.3,4,5 Assim, o tratamento não deve ser protelado em função da espera do resultado do teste ADAMTS13 ou do desenvolvimento de outros sintomas da doença; e a orientação terapêutica (continuar/suspender) deve basear-se na resposta clínica e laboratorial e não no resultado do teste.1,4,5
A duração do tratamento e o número de sessões de plasmafiltração descritos na literatura actual é variável. Algumas guidelines sugerem sessões de 1-1,5 volume de plasma, 1-2 vezes por dia, até 2 dias depois da remissão. Em caso de diagnóstico de PTT hereditária, o tratamento passa pela reposição de ADAMTS13 através de infusão de plasma, na presença de trombocitopenia ou de sintomas, ou de forma profiláctica em intervalos regulares. O uso de corticóides é ainda controverso. Alguns autores consideram fazer parte do tratamento standard5, outros defendem que o seu uso é reservado aos doentes cujo mecanismo imunológico esteja na base da doença.4 No entanto, não existem estudos que comprovem superioridade da associação plasmafiltração com corticóides versus plasmafiltração isoladamente. Numa revisão mais recente de Coppo et al., é descrito que doses mais elevadas de metilprednisolona (10mg/kg/dia 3 dias seguidos de 2,5mg/kg/dia) parecem ser mais eficazes que a dose convencional (1mg/kg/dia).
A idade avançada, valores muito elevados de DHL (reflexo de lesão de órgão) e elevação do nível de troponina cardíaca no momento do diagnóstico estão associados a morte e refractariedade ao tratamento. As recidivas ocorrem em 40% dos doentes e são mais frequentes na deficiência severa de actividade da ADAMTS13 (50%, principalmente no primeiro ano).1,4,6,9-11
Existe uma maior preocupação nas mulheres jovens, dada a associação de PTT com a gravidez.1,4,6 No entanto, o risco de recidiva nesta condição não é claro na literatura, pelo que não existem recomendações quanto à abordagem antes e durante a gravidez na mulher com história de PTT.
O rituximab e outros imunossupressores (ciclofosfamida e vincristina) são usados em casos mais complexos, com resposta subóptima ao tratamento ou exacerbação da doença.5,6,9,10 Estudos com rituximab demonstraram menos e mais tardias recidivas, sugerindo que este deveria ser o tratamento de primeira linha, em associação a plasmafiltração, nos casos de PTT autoimune/adquirida. No entanto, o conhecimento actual sobre o seu uso na PTT é baseado em escassos ensaios, com número limitado de doentes e com nível de evidência moderado.6
No futuro, o desenvolvimento de ADAMTS13 recombinante poderá tornar o tratamento profiláctico mais simples e seguro na PTT hereditária.5 Outros agentes promissores sob investigação incluem a N-acetilcisteína, bortezomib e caplacizumab.6
O Caplacizumab, é um antifactor von Willebrand humanizado, que inibe a interacção entre o os múltimeros do FvW e as plaquetas. Um estudo com 145 doentes com PTT adquirida tratados com caplacizumab versus placebo associado a plasmafiltração mostrou que no grupo do caplacizumab ocorreu normalização mais rápida da contagem de plaquetas; menor taxa de recorrência de PTT; e menor incidência da combinação de morte relacionada com a PTT, recorrência de PTT ou um evento tromboembólico durante o período de tratamento.12
CONCLUSÃO
Este artigo demonstra a importância de um elevado índice de suspeição para o diagnóstico de PTT, perante um doente que se apresenta com anemia e trombocitopenia agudas, com critérios de hemólise microangiopática. Esta apresentação deve ser tratada como uma emergência médica. O atraso no tratamento pode causar danos irreversíveis e a morte.
Quadro I
Microangiopatias trombóticas primárias
| Microangiopatias trombóticas primárias |
| A. Hereditárias |
| 1. Deficiência de ADAMTS13 (PTT congénita) |
| 2. Mediada por alterações do complemento (SHU atípico) |
| 3. Mediada por alterações do metabolismo |
| 4. Mediada por alterações da coagulação |
| B. Adquiridas |
| 5. Deficiência de ADAMTS13 (PTT idiopática) |
| 6. Associada à toxina Shiga (SHU típico) |
| 7. Associada a drogas |
| 7.1. Reacção imune |
| 7.2. Reacção relacionada com a dose tóxica |
| 8. Mediada por alterações do complemento (SHU atípico) |
Tabela 1 - Microangiopatias trombóticas primárias (PTT- púrpura trombocitopénica trombótica; SHU- Síndrome hemolítico urémico)
Quadro II
Microangiopatias trombóticas secundárias
| Microangiopatias trombóticas secundárias |
| Sépsis |
| Neoplasia metastizada |
| Pré-eclampsia severa, eclampsia, síndrome de HELPP |
| HTA grave |
| Doenças auto-imunes (Lúpus, esclerodermia, síndrome antifosfolipídico) |
| Associada ao transplante hematopoiéticos |
Tabela 2 - Microangiopatias trombóticas secundárias (HELPP - Hemolysis Elevated Liver Enzymes Low Platelets; HTA Hipertensão arterial)
Quadro III
Análises à admissão
| Parâmetro analítico | Resultado (valor de referência dos parâmetros alterados) |
| Hemoglobina | 6,4 g/dl (H/M) (ref: 12,0-16,0 g/dL) |
| Plaquetas | <10 000 plaquetas/uL (ref: 150-400 EXP3/uL) |
| Reticulócitos | 8,5% (ref: 0,5-2,5%) |
| Leucograma | Leucócitos: 10700/uL Neutrófilos: 6700/uL Eosinófilos: 200/uL Basófilos: 0/uL Linfócitos: 3000/uL Monócitos: 300/uL |
| Proteína C reactiva | 22,3 mg/L (ref: <3,mg/L) |
| Velocidade de sedimentação | 61 mm/1ªh (ref: 0-20) |
| Creatinina Ureia Sódio Potássio | 0,63 mg/dL 40 mg/dL 138 mEq/L 3,9 mEq/L |
| Exame de urina (sumário e sedimento) | Normal |
| Estudo da coagulação (APTT, PT, INR) | Normal |
| AST Aspartato aminotransferase ALT Alanina aminotransferase Fosfatase Alcalina GamaGT | 61 U/L 27 U/L 64 U/L 16 U/L |
| Esfregaço de sangue periférico | Esquizócitos |
| Desidrogenase láctica | 2009 U/L (ref: 135-225) |
| Bilirrubina total | 6,66mg/dL (ref: <1,2) |
| Bilirrubina directa | 0,98 mg/dL |
| Haptoglobina | <8 mg/dL (ref: 50-320) |
| Teste de Coombs directo | Negativo |
Tabela 3 - Estudo analítico à admissão (H/M hipocrómica/microcítica, APTT - Tempo de tromboplastina parcial activada, TP Tempo de protrombina, INR - razão normalizada internacional)
Quadro IV
Estudo etiológico
| Parâmetro analítico | Resultado (valor de referência dos parâmetros alterados) |
| Beta-HCG | Negativo |
| Hemoculturas (aerobiose e anaerobiose) | Negativas |
| HCV, HIV, HSV 2 e Toxoplasma | Negativos |
| HBV, CMV, Parvovirus B19, HSV 1, EBV | Imune |
| Actividade do complemento CH50 | 249 UA (↑) (ref: 63-145) |
| C3, C4 e C5 | Normais |
| Imunoglobulinas | Normais |
| ANAs, ANCA, Anti dsDNA, FR | Negativos |
| Anticoagulante lúpico, Anti B2-glicoproteína, anti-cardiolipina | Normais |
| TSH/T4 L | Normal |
| Ferro | 293 ug/dL(↑) (ref 49-151) |
| Ferritina | 1486 ng/ml (↑) (ref: 10-120) |
| Ácido fólico | 2,0ng/mL (↓) (ref: 2,2-17,5) |
| Vitamina B12 | 129 pg/mL (↓) (ref: 187-883) |
Tabela 4 Estudo laboratorial efectuado durante o internamento (HCG - Gonadotrofina Coriónica Humana, HIV Vírus da Imunodeficiência Humana, HCV vírus da Hepatite C, HSV 1 e 2 - vírus Herpes Simples 1 e 2, CMV Citomegalovírus, EBV vírus Epstein Barr, ANA Anticorpos Anti-Nuclear, ANCA Anticorpos anti-citoplasma neutrofílico, anti dsDNA - anti double-stranded DNA, FR factor reumatoide, TSH - hormona tireoestimulante, T4 L tiroxina livre)
Quadro V
Apresentação Clínica das PTT
| Apresentação Clínica | Sinal/Sintoma | Alteração laboratorial |
| Anemia Hemolítica Microangiopática | Palidez, astenia, icterícia | Hb: 8-9g/dL Bilirrubina ↑ (+indirecta) LDH ↑ Haptoglobina ↓ Teste Coombs negativo ESP: Esquizócitos (>1%) |
| Trombocitopenia | Petéquias, púrpura, hemorragias | 5 120 000/uL |
| Lesão de órgão | ||
| Envolvimento neurológico | Alterações do estado de consciência/ Défices neurológicos focais (transitórios) | |
| Envolvimento renal | Oligúria/anúria, hematúria | |
| Envolvimento GI | Isquemia intestinal (Dor abdominal, náuseas, vómitos, diarreia) | |
| Envolvimento cardíaco | Arritmia, enfarte agudo do miocárdio, insuficiência cardíaca, morte súbita |
Tabela 5 - Apresentação clínica das PTT (Hb - hemoglobina; LDH - desidrogenase láctica; ESP - esfregaço de sangue periférico)
Figura I
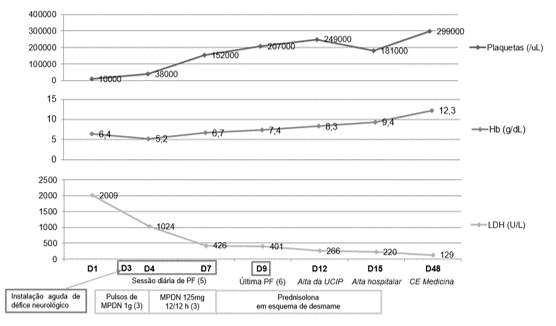
Figura 1- Evolução clínica durante o internamento (MPDN metilprednisolona; PF plasmafiltração; UCIP Unidade Cuidados Intermédios Polivalente; CE Consulta externa)
BIBLIOGRAFIA
(1) George JN. Thrombotic Thrombocytopenic Purpura. N Engl J Med 2006; 354:1927-1935.
(2) Fontana S, Kremer Hovinga JA, Lämmle B, Mansouri Taleghan B. Treatment of thrombotic thrombocytopenic purpura. Vox Sanguinis 2006; 90:245-254.
(3)Scully M, Hunt BJ, Benjamin S, et al. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. British Journal of Haematology 2012; 158(3):323-35.
(4) George JN. How I treat patients with thrombotic thrombocytopenic purpura. Blood 2010; 116(20):4060-9.
(5) George JN, Nester CM. Syndromes of Thrombotic Microangiopathy. N Engl J Med 2014; 371:654-666.
(6) Coppo P. French Reference Center for Thrombotic Microangiopathies. Treatment of autoimmune thrombotic thrombocytopenic purpura in the more severe forms. Transfus Apher Sci. 2017; 56 (1):52-56.
(7) Joly BS, Coppo P,Agnès Veyradier. Thrombotic thrombocytopenic purpura. Blood 2017; 129:2836-2846.
(8) Upadhyay VA, Geisler BP, Sun L, Uhl L, Kaufman RM, Stowell C, Makar RS, Bendapudi PK. Utilizing a PLASMIC score-based approach in the management of suspected immune thrombotic thrombocytopenic purpura: a cost minimization analysis within the Harvard TMA Research Collaborative. Br J Haematol. 2019 May 26. doi: 10.1111/bjh.15932.
(9) Kremer Hovinga JA, Vesely SK, Terrell DR, Lämmle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood 2010; 115:1500-11.
(10) Thejeel B, Garg AX, Clark WF, Liu AR, Iansavichus AV, Hildebrand AM. Long-term outcomes of thrombotic microangiopathy treated with plasma exchange: a systematic review. Am J Hematol 2016; 91:623-30.
(11) Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Clinical importance of ADAMTS13 activity during remission in patients with acquired thrombotic thrombocytopenic purpura. Blood 2016; 128:2175-8.
(12) Scully M, Cataland SR, Peyvandi F, Coppo P, Knöbl P, Kremer Hovinga JA et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N Engl J Med 2019; 380:335-346.