

INTRODUÇÃO
A diarreia é um distúrbio comum, maioritariamente com apresentação aguda, auto-limitada e de etiologia infeciosa. Define-se como diarreia crónica quando se prolonga além das 4 semanas, estimando-se que 1 a 5% da população dos países desenvolvidos serão afetados por pelo menos um episódio ao longo da vida1. O desafio da sua abordagem prende-se com a diversidade de diagnósticos diferenciais. Não obstante, a história clínica e o exame físico detalhados, bem como a seleção criteriosa de exames complementares frequentemente conduzem ao diagnóstico e permitem um plano de tratamento adequado.
CASO 1
Mulher de 74 anos de idade, recorre ao serviço de urgência (SU) por dor abdominal difusa em moedeira, sem fatores de alívio ou agravamento, com 3 meses de evolução e diarreia (3 dejeções por dia) com 3 semanas de evolução. Referia ainda astenia, emagrecimento de 9 Kg, artralgias migratórias intermitentes de caráter inflamatório nos últimos 9 meses e dispneia para pequenos esforços com um mês de evolução. Por este motivo já havia recorrido várias vezes ao SU e realizado diferentes exames complementares de diagnóstico, entre os quais endoscopia digestiva alta que não revelou alterações e tomografia computorizada abdominal que revelou apenas adenopatias retroperitoneais[RM1], as de maiores dimensões com 2 cm de maior eixo. Pela presença dessas adenopatias foi realizada biópsia de gânglio retroperitoneal que levantou suspeita de doença linfoproliferativa, posteriormente excluída por estudo complementar.
Dos antecedentes pessoais destacam-se a implantação depacemakerpor bloqueio aurículo-ventricular completo e doença ulcerosa péptica, submetida a gastrectomia parcial aos 29 anos. Estava medicada com furosemida 40mg id, omeprazol 20mg id e lorazepam 1mg id.
Ao exame objetivo apresentava-se emagrecida (IMC 16,45Kg/m2), com palidez muco-cutânea. A auscultação pulmonar revelava crepitações bibasais e o abdómen era doloroso à palpação profunda dos quadrantes direitos. Apresentava ainda edema discreto no terço inferior dos membros inferiores.
Analiticamente com anemia microcítica e hipocrómica (Hb 9,5g/dL, VGM 78.4fL, CHCM 31.9%), leucocitose (13,28x10^9/L) com neutrofilia (94,8%), proteína C reativa de 12,52 mg/dL e velocidade de sedimentação de 76 mm/h. Ferro sérico e capacidade total de fixação de ferro baixos (Fe 15 µg/dL e CTFF 244 µg/dL), com ferritina e transferrina normais. Proteínas totais séricas diminuídas (4,7 g/dL). Restante estudo analítico sem alterações.
A radiografia do tórax e ecografia abdominal não revelaram alterações relevantes.
Perante a suspeita de uma síndrome de má absorção, repetido o estudo endoscópico, que então revelou duodenite intensa e difusa de toda a mucosa bulbar (Fig. 1). O estudo histológico dos fragmentos de mucosa duodenal revelou a presença de numerosos macrófagos espumosos PAS-positivos (Fig. 2) e o estudo por aplicação de reação polimerase em cadeia foi positiva paraTropheryma whipplei.
Estabelecido o diagnóstico de doença de Whipple foi instituída terapêutica com ceftriaxone 2g endovenoso id, por 2 semanas, posteriormente seguido por sulfametoxazol 800mg/ trimetropim 160mg bid.
Apesar de melhoria clínica e analítica iniciais, um mês após início de tratamento verificada recorrência de diarreia, dor abdominal, emagrecimento e início de vómitos de conteúdo alimentar. Analiticamente apresentava agravamento da anemia (Hgb-8.1g/dL), com persistência de leucocitose e elevação de proteína C reativa. Considerada recidiva da doença, retomando tratamento com ceftriaxone 2g bid EV durante 4 semanas seguido de doxiciclina 100mg bid e hidroxicloroquina 200mg tid durante um ano, ao final do qual se encontrava sem queixas gastrointestinais, com ganho ponderal significativo e sem alterações analíticas.
CASO 2
Mulher de 71 anos recorre ao SU por diarreia com um ano de evolução (3 a 4 dejeções líquidas, diárias, sem sangue ou muco) associado a dor abdominal difusa em cólica. Associadamente apresentava anorexia, enfartamento pós-prandial, perda ponderal de 8Kg, astenia e episódios recorrentes de dor, rigidez e limitação funcional das pequenas articulações das mãos. Por persistência dos sintomas tinha já realizado em ambulatório, endoscopias digestivas alta e baixa, com resultado normal, e TC toraco-abdominal que revelou hepato-esplenomegalia e múltiplas adenomegalias no retroperitoneu e na raiz do mesentério, com diâmetro máximo de 25mm. Por este motivo orientada para consulta de Hematologia sob suspeita de doença linfoproliferativa, que foi excluída.
Dos antecedentes pessoais destaca-se dislipidemia, síndrome depressivo e diagnóstico de trombose venosa profunda três meses antes da admissão, motivo pelo qual se encontrava hipocoagulada com anticumarínico. Estava ainda medicada com sinvastatina 20mg id, mirtazapina 30mg id, zolpidem 10mg id e amilssuprida 50mg id.
No exame objetivo apresentava-se emagrecida (Peso 40.1Kg; IMC 17.1Kg/m2) com palidez mucosa, hiperpigmentação generalizada, hipotensa (TA 89/68mmHg), com abdómen distendido, mole e indolor e edema assimétrico moderado dos membros inferiores (mais exuberante à esquerda).
Do controlo analítico destacou-se anemia multifatorial (Hb 9.3g/dl; VGM 71.6fl; ferro 17ug/ml; CTFF 171ug/dl; saturação da transferrina 10%; ferritina 624.2ng/ml), hipoproteinémia com hipoalbuminémia (proteínas totais 4,1mg/dl e albumina 2,3mg/dl), sem elevação significativa de parâmetros inflamatórios (Leuc. 5.7x10^9/L, PCR 2.71mg/dl e VS 27mm/h).
A radiografia abdominal em ortostatismo (Fig. 3) revelou distensão de ansas intestinais e níveis hidroaéreos, sem presença de ar livre e a radiografia do tórax mostrou derrame pleural bilateral de pequeno volume, sem outras alterações.
Realizada endoscopia digestiva alta e baixa que mostrou pontuado esbranquiçado difuso na mucosa do bulbo e segunda porção duodenais e no íleon terminal, sugestivo de telangiectasias (Fig. 4). O estudo histológico no duodeno e íleo terminal revelou a presença de vilosidades globosas e preenchidas por abundantes macrófagos espumosos com positividade para PAS, mesmo após aplicação de diástase, corroborando o diagnóstico de Doença de Whipple (Fig. 5).
Instituída terapêutica com ceftriaxona 2g id durante duas semanas, com resolução da diarreia, ganho ponderal de 3,7Kg (passou de 40.1Kg para 43.8Kg) em 2 semanas, correcção da hipoalbuminémia, redução do derrame pleural e melhoria da anemia (Hb 10.4g/dl; VGM 74.6fl). Após as duas semanas de terapêutica inicial com ceftriaxona, instituída terapêutica de manutenção com sulfametaxazol + trimetroprim 800/160mg id.
Três semanas após o início da terapêutica de manutenção, reaparecimento de picos febris isolados, astenia e anorexia, sem diarreia. Analiticamente apresentava subida de parâmetros inflamatórios (Leuc. 11.4x10`9/L com predomínio de neutrófilos e PCR 13.23mg/dl) e radiograficamente era agora visível derrame pleural esquerdo de médio volume. A TC tórax excluiu lesão do parênquima pulmonar subjacente. Realizada nova EDA que mostrou mucosa intestinal com aspecto macroscópico normal, mas histologicamente havia persistência de macrófagos PAS positivos. Assumida recidiva de doença pelo que se instituiu esquema terapêutico com ceftriaxona 2g bid durante 4 semanas seguido de esquema de manutenção alternativo com doxiciclina 100mg bid e hidroxicloroquina 200mg tid durante um ano, com resolução completa da sintomatologia, ganho ponderal total de 18Kg (passou de 40.1Kg para 58.1Kg), normalização de parâmetros analíticos e resolução completa do derrame pleural.
Caso 3
Homem de 48 anos referenciado à consulta por picos febris (37.5-38ºC) com dois meses de evolução, cerca de um por semana, associado a perda ponderal significativa (passou de 70kg para 63kg em 2 meses) e sudorese noturna profusa. Tinha já realizado TC abdominopelvica que mostrou hepatomegalia de 20cm, com fígado globoso e de contornos rombos, esplenomegalia ligeira de 134mm e múltiplos e volumosos conglomerados adenopáticos, o maior com 35x16mm. Ao exame físico apresentava hepatoesplenomegalia palpável e adenomegalia duroelástica com cerca de 2 cm na região inguinal direita. Por suspeita de doença linfoproliferativa, realizada biópsia excisional do gânglio inguinal, com resultado histológico suspeito de linfoma T periférico. Avaliado em consulta de Hematologia e após revisão das lâminas excluída doença linfoproliferativa.
Entretanto, teve aparecimento de diarreia, com 3 a 4 dejeções diárias, líquidas, sem sangue ou muco, e dor abdominal em cólica com alívio após a dejeção e associadamente perda ponderal adicional de 5Kg, atingindo peso de 58Kg.
Do estudo complementar destaca-se leucocitose (16.35x10^9/L) com predomínio de neutrófilos (85%), trombocitose (548 000/uL), serologias para hepatite B, hepatite C e VIH não reativos e IGRA negativo. Eletroforese de proteínas sem pico monoclonal e PET que mostrou múltiplas adenomegalias na região abdominal superior e dispersas a nível mesentérico sem hipermetabolismo significativo de 18F-FDG.
A endoscopia digestiva alta não revelou alterações macroscópicas, contudo a histologia da biopsia duodenal evidenciou vilosidades expandidas por infiltrado de macrófagos com citoplasma amplo e vacuolizado e eosinófilos dispersos, o que confirmou a Doença de Whipple. Iniciada antibioterapia com 2g de ceftriaxone diário durante 14 dias, seguido de terapêutica de manutenção com sulfametoxazol-trimetoprim 800/160 bid durante 1 ano, com resolução das queixas e ganho ponderal de 15Kg (passou de 58 para 73Kg).
DISCUSSÃO
A doença de Whipple é uma doença infeciosa, causada pela bactériaTropheryma whipplei, bacilo gram positivo ubiquitário do solo1,2. Consiste numa patologia rara, com uma incidência estimada inferior a 1/1 000 000 e com apenas algumas centenas de casos reportados, maioritariamente na Europa Ocidental e América do Norte3. A doença de Whipple é mais comum em homem brancos, raramente descrita em mulheres (razão masculino-feminino 8-9:1) e usualmente observada na meia idade ou idosos, com exposição ocupacional ao solo3.
A fisiopatologia da doença não está totalmente esclarecida, no entanto considera-se ter transmissão feco-oral com posterior disseminação para os restantes sistemas a partir do epitélio intestinal, macrófagos, capilares e linfáticos1. A maioria dos indivíduos que contacta com oT whippleisão portadores assintomáticos ou sofrem uma infeção limitada pelo desenvolvimento de resposta imunológica3. A infeção crónica é explicada maioritariamente por fatores do hospedeiro, especialmente por predisposição genética: os alelos HLA-DRB1*13 e DQB1*06 foram propostos como predisponentes à patologia4. Pensa-se que a infeção impede a produção de interleucina 12, baixa a produção de interferão-Y pelas células NK e T, alterando a função e ativação de macrófagos. Como resultado, os macrófagos fagocitam as bactérias, mas sendo incapazes de as eliminar e induzir uma resposta imune eficaz, verifica-se acumulação intracelular massiva do microorganismo3-4.
Inicialmente a doença de Whipple foi descrita como uma síndrome de malabsorção, no entanto, é uma doença multissistémica, com atingimento de virtualmente todos os sistemas. A apresentação clássica com artralgias/artrite, febre, dor abdominal e diarreia é rara4. Tipicamente apresenta-se em dois estadios: uma primeira fase ou prodrómica com sintomas constitucionais e queixas articulares e, uma segunda fase ou estadio gastrointestinal, em que as queixas digestivas são predominantes1.
Os sintomas articulares estão presentes em cerca de 80% dos doentes e são a primeira manifestação tipicamente com artralgias migratórias das grandes articulações, sendo também possível a ocorrência de oligo ou poliartrite migratória crónica, a mimetizar doença reumatológica5. Nos casos em que as queixas articulares são a manifestação inicial, os sintomas gastrointestinais surgem em média em 6 anos, levantando a suspeita diagnóstica5. Na maioria das vezes ocorre diarreia intermitente associada a dor abdominal, embora esteatorreia e hemorragia gastrointestinal oculta também estejam descritas. Ao longo da progressão da doença, estes processos culminam em síndrome consumptivo e perda de peso. A febre não se encontra presente de forma universal, sendo reportada em apenas 25 a 45% dos casos1-5. Em cerca de metade dos casos estão presentes adenomegalias mesentéricas e mediastínicas, podendo levantar a suspeita de doença linfoproliferativa1. O envolvimento cardíaco ocorre em aproximadamente 30% dos doentes e a doença de Whipple constitui a principal etiologia da endocardite com hemoculturas negativas4,6.
Ao exame físico, os doentes podem apresentar muitos dos estigmas associados à malabsorção, sendo no entanto inespecíficos da condição: caquexia, hiperpigmentação periorbital e malabsorção associada ao défice de vitaminas D e B12, glossite, queilite angular, gengivite ou hemorragia gengival, bem como distensão abdominal e cegueira nocturna associada ao défice de vitamina A1. Podem ainda ser encontrados sinais de atingimento articular na forma de artrite, sacroileíte ou estreitamento do canal cárpico, no entanto a deformidade articular é rara1,5. O envolvimento do SNC, descrito em cerca de 20 a 40% dos doentes, é mais frequentemente assintomático e apenas diagnosticado por técnicas moleculares no líquido cefalorraquidiano (LCR)7. Contudo pode manifestar-se por défice de memória, confusão mental e síndrome demencial e mais raramente pode ocorrer ataxia cerebelosa, hemiparesia, neuropatia periférica ou convulsões4. A presença de miorritmia oculomastigatória (movimentos de convergência ocular associados à contração de músculos mastigatórios) e miorritmia oculo-facial-esquelética ocorrem em cerca de 20% dos doentes com atingimento central e são considerados patognomónicos da doença4,7.
O diagnóstico de doença de Whipple deve ser suspeitado em doentes com as manifestações cardinais, após exclusão de outras patologias mais frequentes que cursam com diarreia crónica. O diagnóstico torna-se mais complexo nos doentes que não desenvolvem sintomas gastrointestinais, podendo ser suspeitada em doentes com poliartrite migratória seronegativa que não responde a terapêutica imunossupressora ou doentes com febre de origem desconhecida.
No estudo complementar, os exames laboratoriais frequentemente sugerem a presença de malabsorção com a presença de anemia, hipoalbuminemia e prolongamento do tempo de protrombina (secundário ao défice de vitamina K). Frequentemente estão presentes leucocitose, trombocitose e elevação da proteína C reativa1. O teste definitivo para a presença de malabsorção é a determinação da gordura fecal, anormal se superior a 6g/24h, não sendo contudo específica para doença. Os exames imagiológicos poderão mostrar envolvimento pleuropulmonar como derrame pleural, doença pulmonar intersticial e hipertensão pulmonar4,9.
Nos casos suspeitos deve ser realizada endoscopia digestiva alta com biópsias de intestino delgado para coloração por PAS, imunohistoquimica e PCR. Se inconclusivas, devem ser colhidas amostras de outros locais envolvidos (liquido sinovial, gânglios linfáticos, LCR). No caso de não existirem sintomas gastrointestinais, as amostras devem ser colhidas nos locais anatómicos relevantes e caso estas amostras sejam indeterminadas, e se mantenha a suspeita clínica, deve ser realizada biópsia do intestino delgado para esclarecimento diagnóstico1.
Na maioria dos casos, a análise macroscópica da biópsia de intestino delgado não demonstra alterações de relevo, mas ocasionalmente apresenta mucosa pálida, espessada e com exsudato fibroso na superfície peritoneal1,4. A avaliação microscópica permite o diagnóstico na presença dos achados típicos: atrofia de vilosidades, distensão da mucosa por macrófagos espumosos PAS positivos, bem como extenso material PAS positivo na lamina própria e que se extende àmuscularis mucosae submucosa, com coloração Ziehl-Neelsen negativa1,3,5. Estes macrófagos PAS positivos podem ainda ser encontrados noutros tecidos, nomeadamente gânglios linfáticos periféricos e mesentéricos. Na ausência destes achados o diagnóstico pode ser confirmado pela positividade de dois testes diferentes (PAS, PCR ou imunohistoquimica) na mesma amostra ou dois testes paraT. whippleiem amostras diferentes. Se apenas um teste é positivo, a doença de Whipple é possível no entanto o diagnóstico mantem-se incerto1. O diagnóstico de endocardite porT. Whippleigeralmente é confirmado por histologia ou imunohistoquimica de uma válvula ressecada6. A pesquisa de ADN bacteriano pela técnica de PCR, quando de forma isolada, possui elevada taxa de falsos positivos que podem decorrer de colonização assintomática ou pela presença de bactérias estreitamente relacionadas8. A pesquisa de anticorpos IgG não deve ser usada para o diagnóstico da doença já que se encontra presente em cerca de 70% dos controlos saudáveis e o anticorpo IgM, apesar de mais específico, encontra-se tecnicamente menos disponível9.
A terapêutica principal da doença de Whipple é a antibioterapia, cujo objetivo é a redução da morbilidade, prevenção das complicações e erradicação da infeção. Dada a elevada frequência de recidiva associada a cursos de antibioterapia curtos, preconiza-se terapêutica com duração prolongada. A terapêutica padrão inicial é ceftriaxona 2g id intravenosa ou 2 milhões de unidades (MU) de penicilina G a cada 4h durante 14 dias. Após o tratamento inicial, institui-se o tratamento de manutenção com trimetoprim-sulfametoxazol 160/800mg oral durante um ano1,4. Se evidencia de endocardite ou envolvimento do SNC, alguns especialistas defendem uma terapêutica endovenosa inicial durante 4 semanas, seguida de terapêutica de manutenção oral durante 1 ano7. Nos casos de doença do SNC sugere-se que a dose de ceftriaxona seja 2g 12/12h durante quatro semanas. No caso de doentes com alergia aos beta-lactâmicos, o esquema alternativo na fase inicial é meropenem 1g a cada 8h, durante 2 a 4 semanas, seguido da terapêutica de manutenção4. No caso de alergia aos compostos sulfa, o esquema alternativo para a fase de manutenção é a associação de doxiciclina 100mg oral bid e hidroxicloroquina 200mg oral tid1.
A avaliação da resposta ao tratamento pode ser realizada através da avaliação da sintomatologia, peso e hematócrito. Não existem estudos suficientes para sugerir repetição de testes para pesquisa deT. whipplei, embora frequentemente se repita a biópsia de intestino delgado anualmente durante os primeiros 5 anos e posteriormente cada 3 a 5 anos. Para doentes com PCR positiva no LCR, a pesquisa pela técnica molecular deve ser repetida a cada 2 a 3 meses até negatividade4,7.
Em cerca de 17 a 35% dos casos não ocorre a melhoria esperada após início da terapêutica, ou há recorrência dos sintomas após melhoria inicial10. Nestas situações podem ser utilizados esquemas alternativos como terapêutica inicial com ceftriaxona 2g iv 12/12h ou 4 MU de penicilina G 4/4h durante 4 semanas seguido de terapêutica de manutenção com duração de um ano com TMP-SMZ 960mg 12/12h ou esquema combinado de doxiciclina 100mg oral bid e hidroxicloroquina 200mg oral tid.
O prognóstico da doença de Whipple sem tratamento é reservado e a mortalidade aproxima-se de 100% um ano após início de síndrome consumptivo1. Se a terapêutica recomendada é cumprida, o prognóstico é bom, com remissão clínica em cerca de 70% dos doentes durante o primeiro curso de terapêutica, apesar de taxas de recidiva de cerca de 30 a 40%, sobretudo se atingimento do SNC4,5,7.
Figura I
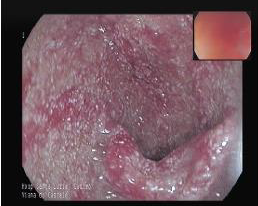
Bulbo duodenal com inflamação intensa e difusa de toda a mucosa.
Figura II
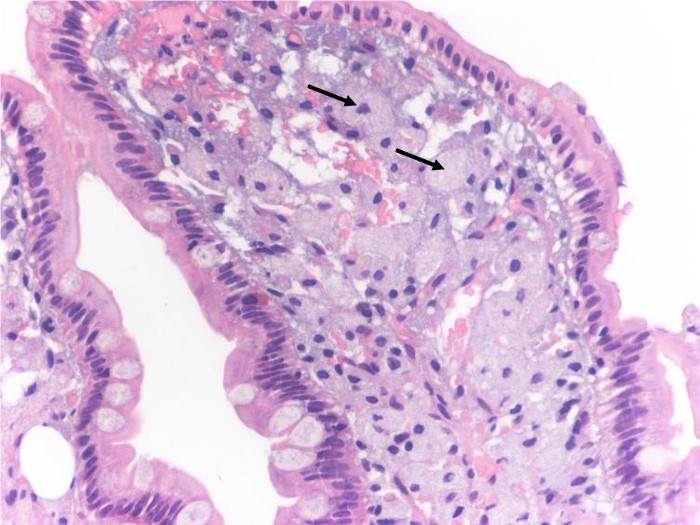
A. Hematoxilina-eosina. Vilosidade duodenal com expansão da lâmina própria por numerosos macrófagos espumosos (setas).
Figura II
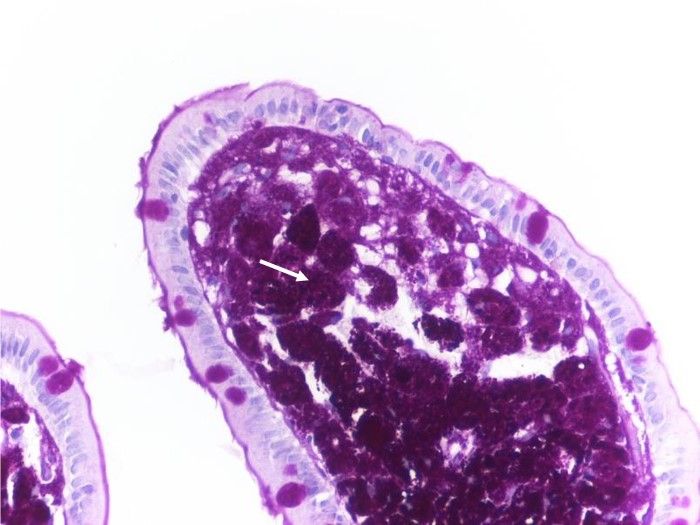
B. Ácido periódico de Shiff. Vilosidade duodenal preenchida com numerosos macrófagos PAS-positivos (seta).
Figura III
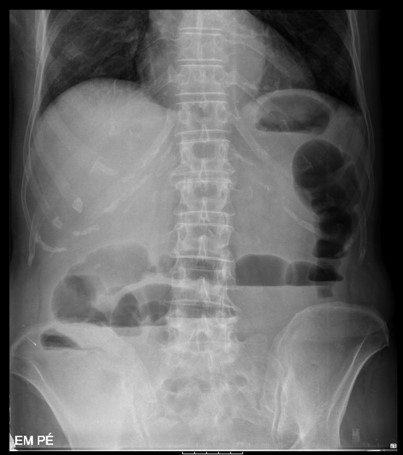
Radiografia abdominal em ortostatismo onde é possível observar os níveis hidroaéreos.
Figura IV
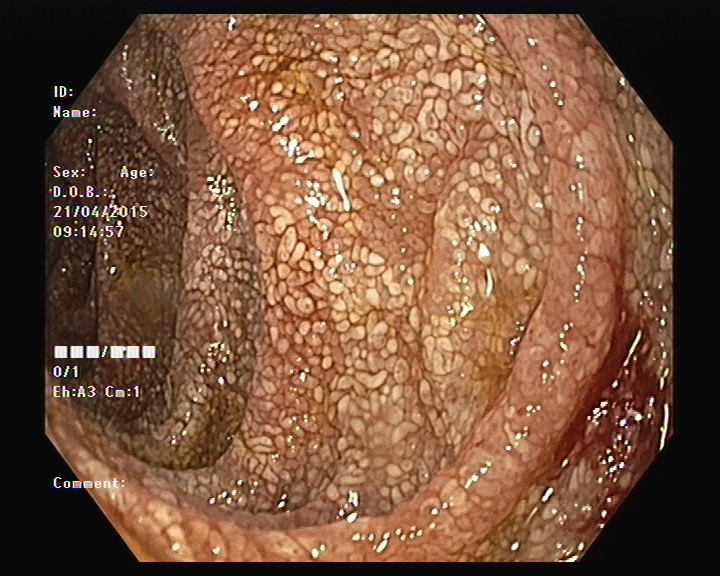
Telangiectasias duodenais.
Figura V
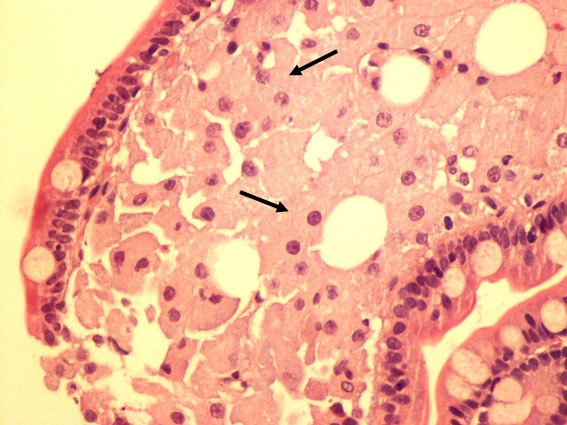
Hematoxilina-eosina. Vilosidades globosas e preenchidas por abundantes macrófagos espumosos (setas).
BIBLIOGRAFIA
1. Fenollar FM, Puèchel X, Raoult D. Whipples Disease. Engl J Med 2007;356:55-66
2. Shiller L, Pardi D, Sellin J. Chronic Diarrhea: Diagnosis and Management.Clin Gastroenterol Hepatol.2017 Feb;15(2):182-193.e3
3. Desnues B, Ihrig M, Raoult D, Mege J, Whipples Disease: a Macrophage Disease. CLINICAL AND VACCINE IMMUNOLOGY, Feb. 2006, p. 170178 Vol. 13, No. 2
4. Marth T. Tropheryma whipplei, Immunosuppression and Whipples Disease: From a Low-Pathogenic, Environmental Infectious Organism to a Rare, Multifaceted Inflammatory Complex. Dig Dis 2015;33:190199
5. Puéchal X. Whipples arthritis.Joint Bone Spine Volume 83, Issue 6, December 2016, Pages 631-6356. Herrmann M, Neumayr A, Essig A, Spiess J, Merk J, Mcoller P. Isolated Whipples Endocarditis. An Underestimated Diagnosis That Requires Molecular Analysis of Surgical Material. Ann Thorac Surg 2014;98:e13)
7. Gerard A, Sarrot-Reynauld F, Liozon E, Cathebras P, Besso G, Robin C et al. Neurologic Presentation of Whipple Disease. Medicine (Baltimore).2002 Nov;81(6):443-57
8. Ramzan N, Loftus E, Burgart L, Rooney M, Batts K, Wiesner R et al. Diagnosis and Monitoring of Whipple Disease by Polymerase Chain Reaction. Ann Intern Med. 1997;126:520-527
9. Dutly F, Altewegg M. Whipples Disease And Tropheryma Whippelii.Clin Microbiol Rev.2001 Jul;14(3):561-83
10. Durand D, Lecont C, Cathebras P, Rousset H, Godeau P. Whipple Disease. Clinical review of 52 cases. Medicine:May 1997; Volume 76; Issue 3:p 170-184