INTRODUÇÃO
A doença de Rendu Osler Weber (DROW), ou Telangiectasia Hemorrágica Hereditária, é uma doença autossómica dominante rara, com prevalência estimada de 1-2 casos em 10 000 pessoas, que foi inicialmente descrita na segunda metade do século XIX, recebendo a designação de DROW já no início do século XX.1,2 É caracterizada por angiodisplasias de atingimento muco-cutâneo e/ou visceral (nomeadamente pulmonar, hepática, do trato gastrointestinal e sistema nervoso central), condicionando hemorragias recorrentes.
O envolvimento hepático pela DROW é considerado relativamente frequente, embora raramente sintomático e, das apresentações clínicas possíveis, a encefalopatia hepática (EH) é extremamente rara.1,3
CASO CLÍNICO
Homem de 70 anos com antecedentes patológicos de DROW tipo 2 diagnosticada 5 anos antes (mutação no gene ACVRL 1 - Exão 6, c.743_744delCA, p.Thr248Serfs*143), prostatectomia por neoplasia maligna, doença renal crónica sob hemodiálise, insuficiência cardíaca crónica com cardiopatia isquémica, fibrilhação auricular, hipertensão arterial e dislipidemia. Foi admitido no Serviço de Urgência (SU) por desorientação temporo-espacial com uma hora de evolução. De acordo com a esposa, o doente apresentava episódios semelhantes nos últimos dois meses. No exame objetivo destacava-se desorientação temporo-espacial, discurso lentificado, auscultação cardíaca arrítmica e flapping. Relativamente ao estudo analítico realizado, na Tabela 1 encontram-se descritos os resultados mais relevantes.
Foi realizada tomografia computorizada (TC) cranioencefálica que não demonstrava lesões isquémicas, hemorrágicas ou lesões ocupando espaço. A radiografia do tórax também não apresentava alterações de relevo. Durante a vigilância, por novo episódio de agitação e dor abdominal, foi pedida TC do abdómen. Esta mostrava fígado com textura heterogénea sugestivo de hepatopatia crónica, com vários esboços nodulares na periferia do parênquima, relacionados com vasculatura?, baço ligeiramente globoso e pequeno volume de ascite. Foi então, realizada elastografia hepática (Fibroscan®) com valor de 45,6 kPa, compatível com cirrose hepática. Este exame foi efetuado mais de 24 horas após a última sessão de hemodiálise.
Admitiu-se o diagnóstico de cirrose hepática de causa ainda não identificada, apresentando-se como EH de tipo C, tendo sido iniciada terapêutica com rifaximina 1200mg por dia e lactulose três vezes por dia.
No decurso da investigação etiológica, foi realizada TC abdominal contrastada trifásica mostrando fígado com bordos regulares, aumento do calibre da veia porta (17mm) e da artéria hepática (7mm), hipertrofia do lobo caudado e múltiplos shunts portovenosos (Fig. 1). Dada a discordância entre os resultados das imagens de TC abdominal e da elastografia, optou-se pela realização de biópsia hepática, guiada por exame de imagem, que revelou parênquima sem cirrose, com alterações vasculares enquadráveis com envolvimento hepático por DROW (Fig. 2).
Foi então assumido, como diagnóstico definitivo EH de tipo B como primeira manifestação de envolvimento hepático por DROW.
O doente tem tido múltiplas vindas ao SU por EH grau III-IV apesar de terapêutica médica sintomática otimizada, estando a ser avaliada a sua elegibilidade para transplante hepático ou transplante duplo hepático e renal.
DISCUSSÃO
A DROW é uma entidade rara cujo diagnóstico clínico é baseado na presença de pelo menos três dos seguintes achados clínicos: epistaxis recorrentes, telangiectasias cutâneas ou mucosas (jugal e/ou intranasal), envolvimento visceral (pulmão, fígado, trato gastrointestinal e sistema nervoso central) e história familiar de DROW.3
O envolvimento hepático pela DROW é considerado relativamente frequente (8-31% em estudos retrospetivos e 41-78% em estudos prospetivos),1,4 embora raramente sintomático, os achados mais referidos são os relacionados com insuficiência cardíaca de alto débito, hipertensão portal e doença biliar.1Extremamente raros são os casos clínicos com EH como manifestação do envolvimento hepático na DROW,1,3 não estando nenhum caso publicado em Portugal e apenas 19 na literatura mundial.5
Na maioria dos casos, a DROW é causada por mutações em um de dois genes: gene codificador da engoglina (ENG), associada à DROW tipo 1, ou o gene codificador da activin receptor-like kinase type 1 (ALK-1 ou ACVRL1), associada à DROW tipo 2. Em alguns casos estas mutações não são detetadas, pelo que se coloca a hipótese de existir uma outra mutação num terceiro gene.1 O envolvimento hepático pela DROW está mais frequentemente descrito na DROW tipo 2.6
No caso clínico descrito, existiram vários fatores confundidores na marcha diagnóstica. Inicialmente foram averiguadas como possíveis etiologias do quadro de desorientação temporo-espacial síndrome urémico, lesão cerebrovascular ou processo infecioso. De salientar que, para exclusão de foco infecioso, foram determinantes a ausência de sinais meníngeos ou outros sinais focalizadores ao exame objetivo (excetuando o quadro de desorientação) sugestivos de infeção, a ausência de critérios clínicos de síndrome de resposta inflamatória sistémica e o facto de os exames de imagem e de urina não apresentarem alterações compatíveis com infeção.
Excluídas estas etiologias e associando os achados clínicos às alterações analíticas, elevação da enzimologia hepática e da amoniémia, com as alterações na TC abdominal sugestivas de hepatopatia crónica, foi assumido diagnóstico de EH de causa a esclarecer.
Realizada a elastografia hepática (fibroscan®), o valor obtido foi compatível com cirrose hepática. Deste modo, considerou-se que a EH tinha como causa uma cirrose hepática, embora a TC abdominal não descrevesse imagem da habitual dismorfia do parênquima hepático, mais típica de cirrose estabelecida. Fazia ainda referência a esboços nodulares, questionando poderem corresponder a estruturas vasculares.
A TC trifásica com contraste foi esclarecedora quanto à presença de estruturas vasculares anómalas no parênquima hepático, características do envolvimento hepático pela DROW: a dilatação da veia porta e da artéria hepática e a presença de múltiplos shunts portovenosos em todo o parênquima.7 A biópsia hepática além de identificar a presença de vasos anómalos, também mostrou que o parênquima hepático não tinha cirrose.
Tendo em conta os achados clínicos, as alterações imagiológicas (dilatação da artéria hepática e da veia porta e os múltiplos shunts portovenosos dispersos por todo o parênquima hepático), a biópsia hepática e a associação mais frequente da DROW tipo 2 ao envolvimento hepático por esta doença, foi possível assumir o diagnóstico final de EH não em contexto de cirrose, mas sim de envolvimento hepático por DROW.
Os valores da elastografia hepática obtidos, compatíveis com cirrose, podem ser justificados pela relação temporal do fibroscan® com a última sessão de hemodiálise. Em vários estudos realizados, foi demonstrada uma grande variação nos valores da elastografia hepática, relacionada com o intervalo temporal entre o exame e a sessão de hemodiálise, sendo mais fidedigno o valor obtido logo após a sessão. Assim, a sobrecarga de volume no doente com doença renal crónica já sob TSFR, parece influenciar os valores obtidos, nomeadamente sobrevalorizar os mesmos em doentes sem fibrose hepática ou com fibrose ligeira. No doente apresentado, a elastografia foi realizada mais de 24 horas após a sessão de hemodiálise.8
A abordagem terapêutica na DROW tem como principal objetivo o controlo sintomático e prevenção das complicações associadas, nomeadamente as hemorrágicas. O tratamento dirigido no envolvimento hepático pelo DROW está preconizado apenas quando este é sintomático. Nos doentes que apresentam EH, a abordagem tende a ser mais complexa.
Nos doentes com envolvimento hepático sintomático por DROW, o objetivo terapêutico passa pelo controlo dos sintomas e tentativa de redução dos shunts existentes, nomeadamente pelo uso de fármacos antiangiogénicos e/ou embolização/ligação mecânica da artéria hepática.7 O único tratamento com intenção curativa é o transplante hepático, embora na literatura sejam referidas, com alguma frequência, complicações hemorrágicas associadas ao procedimento cirúrgico, estando também identificada a possibilidade de recorrência após o transplante.1,7 Quando nem o transplante hepático, nem a redução dos shunts portovenosos são possíveis, o tratamento médico sintomático da EH deverá ser semelhante ao utilizado nos doentes com cirrose hepática avançada.1
No doente apresentado, foi iniciada terapêutica médica conservadora otimizada da EH, com ligeira melhoria inicial, mas ineficaz a médio prazo dada a persistência dos sintomas (Fig. 3).
O doente foi proposto para transplante hepático e renal, estando a ser avaliada a sua elegibilidade para o transplante. Tendo em conta todas as comorbilidades do doente, será questionada a sua elegibilidade, pelo que poderão ser alternativas de segunda linha a embolização mecânica de shunts portovenosos ou o início de fármaco antiangiogénico, nomeadamente o bevacizumab. Este fármaco tem sido utilizado em vários doentes com DROW com envolvimento hepático complicado de doença cardíaca de alto débito, com resultados promissores em termos de sintomatologia e qualidade de vida nestes doentes.9 Num estudo que avaliou a redução imagiológica dos shunts portovenosos com o uso de bevacizumab, não pareceu existir evidência de redução significativa do seu número.10 Mais estudos serão necessários, nomeadamente em doentes com EH por envolvimento hepático por DROW para avaliação destes dados.
CONCLUSÃO
Apresentamos este caso clínico pela sua raridade e pela complexidade da estratégia diagnóstica, salientado que apesar da encefalopatia por envolvimento hepático da DROW ser extremamente rara, deve ser excluída num doente com história de DROW e alteração do estado de consciência, mesmo sem conhecimento prévio de envolvimento hepático pela doença.
AGRADECIMENTOS
Os autores agradecem a colaboração do Dr. Rui Caetano Oliveira do Serviço de Anatomia Patológica do Centro Hospitalar e Universitário de Coimbra pela cedência das imagens histológicas e sua ajuda na interpretação das mesmas.
PRÉMIO
Este caso clínico foi apresentado como Poster nas XIII Jornadas do Núcleo de Estudos das Doenças do Fígado da Sociedade Portuguesa de Medicina Interna, decorridas no Porto a 4 e 5 de outubro de 2019, tendo sido atribuído Prémio Menção Honrosa.
Quadro I
Resultados a salientar do estudo analítico realizado
| Parâmetro analítico | Resultado |
| | |
| Hemoglobina | 8.9 g/dL * |
| Leucócitos | 5 250/µL |
| Proteina C reativa | 13.2 mg/dL |
| Alanina aminotransferase (ALT) | 51 U/L (2 x LSN**) |
| Lactato desidrogenase (LDH) | 708 U/L (1.75 x LSN **) |
| Fosfatase alcalina (FA) | 182 U/L (1.5 x LSN **) |
| ỿ-glutamil transferase (GGT) | 84 U/L (1.5 x LSN **) |
| Bilirrubina total | 2.58 mg/dL (2.5 x LSN **) |
| Amoniémia | 234.7 µg/dL (2.75 x LSN **) |
| Azotémia | 19.7 mg/dL |
| Etanol sérico | Negativo |
| Exame sumário de urina | Não sugestivo de infeção do trato urinário |
| Pesquisa de drogas de abuso na urina | Negativa |
* Anemia normocítica/normocrómica já conhecida e dentro dos valores habituaisdo doente; ** LSN limite superior do normal-
Figura I
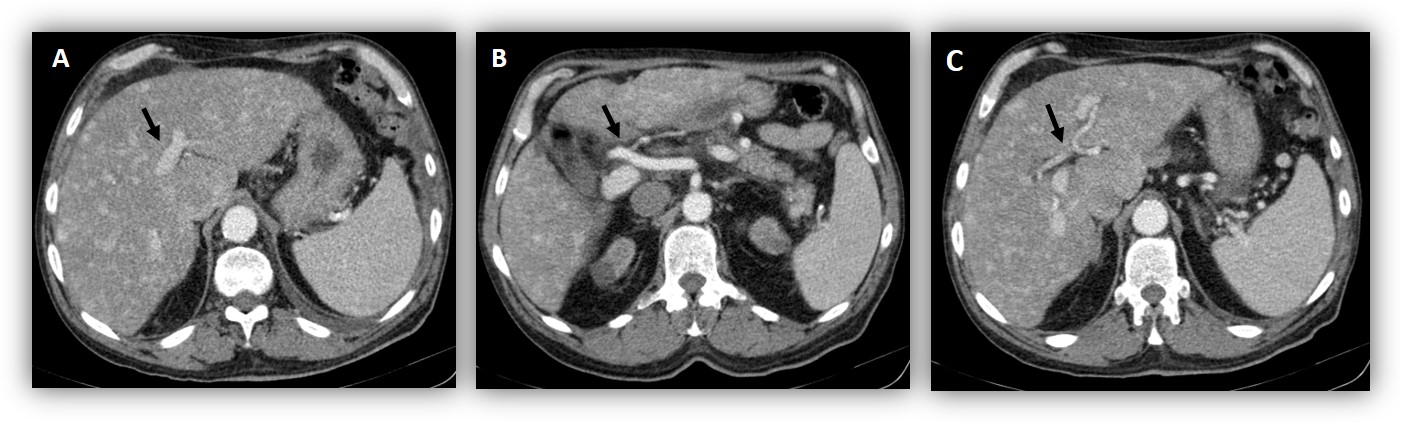
TC abdominal com contraste trifásica - Aumento do calibre da veia porta (17mm) e da artéria hepática (7mm), hipertrofia do lobo caudado e múltiplos shunts portovenosos. A e B setas a indiciar a veia porta dilatada (17 mm de diâmetro máximo); C - seta a indicar a artéria hepática dilatada (7mm de diâmetro máximo) com bifurcação nos ramos esquerdo e direito.
Figura II
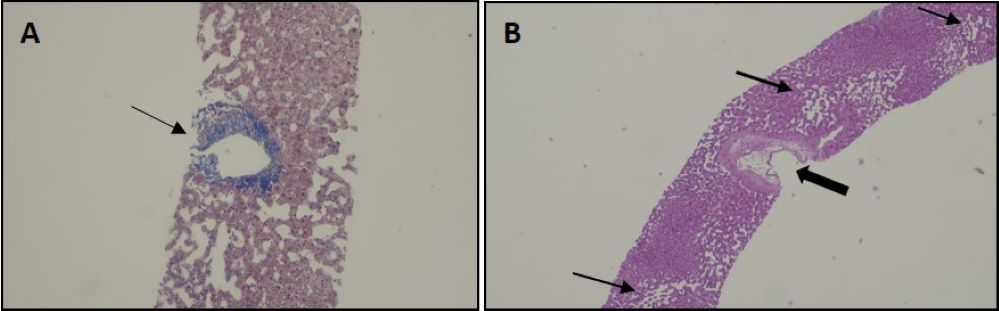
Imagens histológicas dos fragmentos obtidos na biópsia hepática. A - Vaso sanguíneo anómalo com alterações fibróticas (seta); coloração com Tricrómio de Masson, ampliação 100x. B - Dilatação sinusoidal (setas estreitas), com vaso sanguíneo anómalo e com alterações da parede (seta larga); coloração com Hematoxilina & Eosina, ampliação 40x.
Figura III
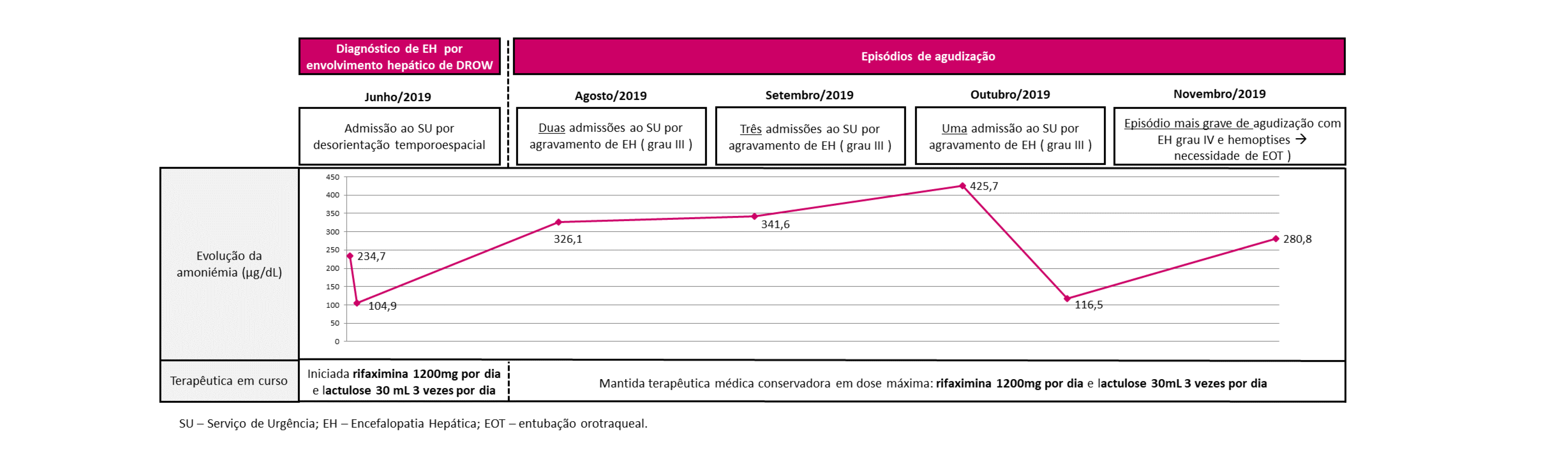
Evolução temporal clínica, com quantificação e caracterização dos episódios de agudização, evolução dos valores de amoniémia e terapêutica efetuada.
BIBLIOGRAFIA
1- Garcia-Tsao, G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT) Journal of Heatology 2007; 46: 499507. doi:10.1016/j.jhep.2006.12.008.
2- Sadick H., Sadick M., Hörmann K. Neurocutaneous Disorders Phakomatoses and Hamartoneoplastic Syndromes. 2008. Chapter 11, p. 311-21.
3- Ha J., Kwan Son B., Bong A., Kwan Jo Y., Kim S., Jo Y., Park Y., Jung Y. Osler-Weber-Rendu Disease Presenting as Recurrent Portosystemic Encephalopathy in a 75-year-old Female Patient. Korean J Gastroenterol 2015;65:57-61. doi: 10.4166/kjg.2015.65.1.57.
4- Piskorz, M., Waldbaum, C., Volpacchio, M., Sordá, J. Liver involvement in hereditary hemorrhagic telangiectasia. Acta Gastroenterol Latinoam 2011; 41:225‑9.
5- Ncbi.nlm.nih.gov pubmed [homepage na Internet]. PubMed searching hepatic+encephalopathy+rendu+osler+weber+case+report; 2020 (consultado em 15 Jan 2020). Disponível em: //www.ncbi.nlm.nih.gov/pubmed/?term=hepatic+encephalopathy%2C+rendu+osler+weber%2C+case+report
6- Bossler A.D., Richards J., George C., Godmilow L., Ganguly A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagictelangiectasia (HHT): correlation of genotype withphenotype. Hum Mutat 2006; 27:667-75. doi: 10.1002/humu.20342.
7- Pinto E.; Lourenço L.;Costa A. Envolvimento hepático na telangiectasia hemorrágica hereditária. RevClinHosp Prof Dr Fernando Fonseca 2013; 2(1): 47-51.
8- Caragea, D. C., Ungureanu, B. S., Florescu, D. N., Popa, P., Sacerdotianu, M. V., Gheonea, D. I., et al. Noninvasive Fibrosis Assessment in Chronic Viral Hepatitis C associated with End Stage Renal Disease. Current health sciences journal, 2018; 44(3), 206210. doi: 10.1007/s00261-018-1671-4.
9- Chavan A., Schumann-Binarsch S., Schmuck B., Oltmer F., Geisthoff U., Hoppe F., et al. Emerging role of bevacizumab in management of patients with symptomatic hepatic involvement in Hereditary Hemorrhagic Telangiectasia. Am J Hematol. 2017 Nov;92(11): E641-E644.
10- Dupuis-Girod S., Ginon I., Saurin J.C., Marion D., Guillot E., Decullier E., et al. Bevacizumab in Patients With Hereditary Hemorrhagic Telangiectasia and Severe Hepatic Vascular Malformations and High Cardiac Output. JAMA. 2012 Mar 7;307(9):948-55.

