Mulher de 52 anos recorre ao SU por toracalgia pós-traumática. Doente referia concomitantemente cansaço para pequenos esforços com meses de evolução e agravamento recente. Como antecedentes relevantes destaca-se apenas a descrição de múltiplos episódios de epistáxis espontâneas e recorrentes.
À observação inicial doente encontrava-se apirética e eupneica, observando-se mucosas acianóticas com múltiplas telangiectasias nos sulcos nasogenianos, palato e lábios; auscultação pulmonar e cardíaca sem alterações.
A doente apresentava saturações periféricas de 87% em ar ambiente, correspondendo a uma PaO2 50mmHg avaliada na posição sentada, ortodesoxia não foi excluída. Tal avaliação equivale a um gradiente alveolar-arterial de 60.4mmHg (calculado para um quociente respiratório estimado de 0.8).
Analiticamente destaca-se apenas policitémia (Hb18g/dL; Htc 58,4%), sem elevação dos parâmetros inflamatórios.
O estudo imagiológico inicial por radiografia de toráx revelou hipotransparência da base pulmonar direita (Fig. 1), descrita posteriormente em Angio TC torácica como malformação arteriovenosa (MAV) com ingurgitamento evidente de uma das veias pulmonares direitas e ramo lobar inferior da artéria pulmonar direita, e densificação do parênquima adjacente na porção distal destes ramos vasculares (Fig. 2).
O ecocardiograma TT excluiu shunt direito-esquerdo intracardíaco, apresentando baixa probabilidade de hipertensão pulmonar (HTP).
Optou-se assim pela realização de flebotomia, com subsequente descida do hematócrito para 46,2%, e a doente teve alta sob oxigenioterapia e antiagregação. Por se tratar de uma hipoxemia sintomática, com fração de shunt inferida >5% e risco de embolização paradoxal a doente foi orientada para exclusão da MAV.
Avaliada em consulta de Radiologia de Intervenção, constatou-se que dado o risco inerente de HTP associado à embolectomia de grandes dimensões e elevada probabilidade de persistência da MAV tornava a doente melhor candidata para lobectomia inferior direita, tendo sido submetida a este procedimento cerca de 1 mês depois. Tal decorreu sem intercorrências major, tendo sido descritas uma miríade de alterações vasculares à superfície dos pulmão na forma de telangiectasias; pós operatório sem necessidade de oxigenioterapia suplementar com manutenção de SpO2>95%.
Doente manteve seguimento em consulta de Medicina Interna onde se optou por realizar estudo complementar para exclusão de outras MAV. Considerou-se que as telangiectasias descritas, epistáxis recorrentes e MAV pulmonar identificada permitiam, segundo os critérios Curaçao1 o diagnóstico de telangiectasia hemorrágica hereditária, mesmo na ausência de história familiar conhecida.
Esta síndrome autossómica dominante é associada à mutação dos genes ENG e ACVRL1 que codificam recetores TGF-ß, associado à dilatação e eventual alteração da vasculatura.2,3 No caso da nossa doente o estudo genético destes por MLPA (Amplificação Multiplex de Sondas Dependente de Ligação) não identificou grandes deleções ou duplicações. No entanto, há que considerar que 90% das alterações nestes genes são mutações pontuais ou deleções/inserções não detetáveis por esta técnica.4Por este motivo, optou-se pela realização de angio TC-CE , estudo endoscópico gastrointestinal e doppler abdominal; a reavaliar posteriormente eventual necessidade de análise genética por sequenciação.
As MAV pulmonares são raras mas um diagnóstico diferencial de problemas clínicos comuns. São mais comummente múltiplas, de tamanho, número e distribuição variável embora descritas maioritariamente nos lobos inferiores.5 Apesar de muitas destas serem congénitas, a idade média de apresentação é geralmente após a 4ª década de vida, sendo que são 1.5 a 2 vezes mais comuns no sexo feminino.6
Figura I
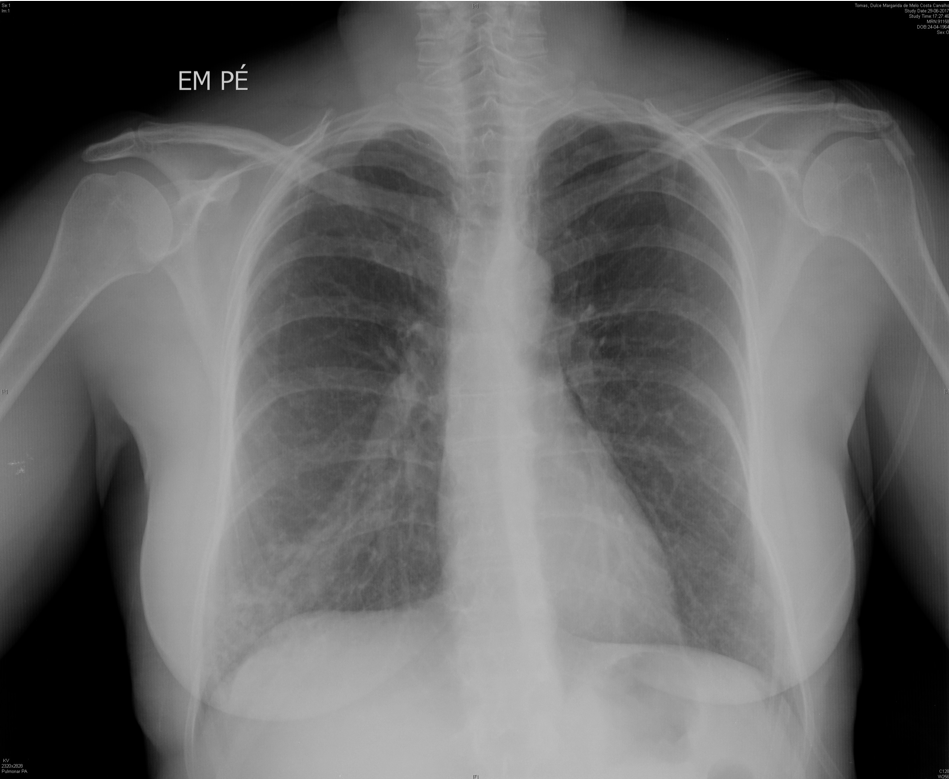
Rx toráx - hipotransparência no lobo inferior pulmonar direito
Figura II
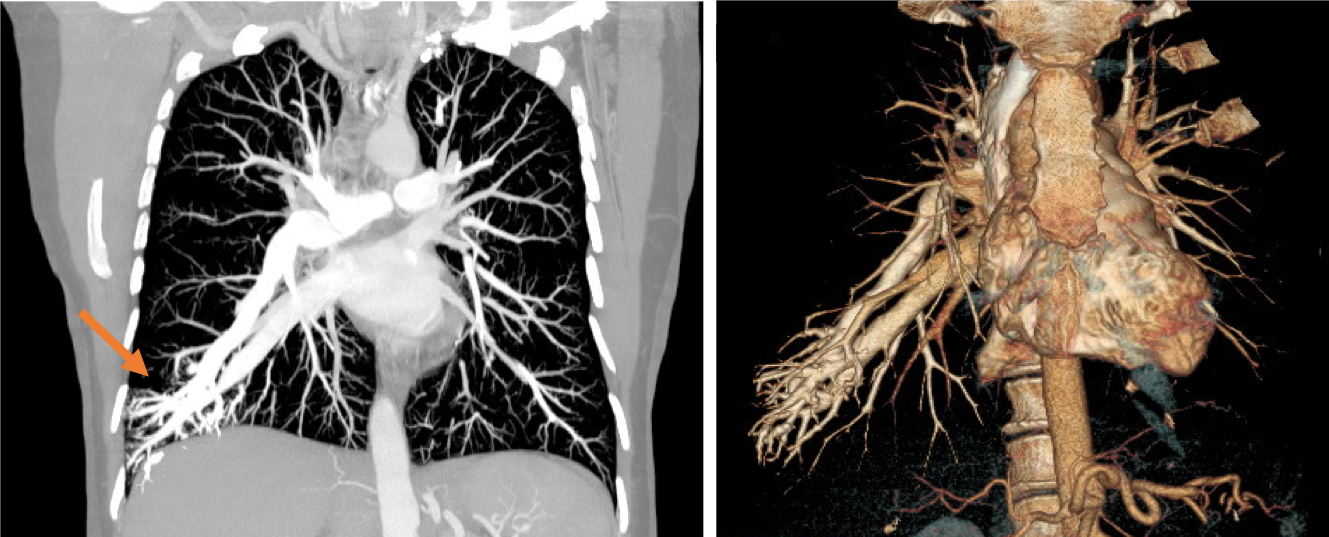
AngioTC torácica e reconstrução - Ingurgitamento de veias pulmonares direitas e do ramo lobar inferior da artéria pulmonar direita.
BIBLIOGRAFIA
1. Faughnan ME, Palda VA, Garcia-Tsao G, et al.International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. Journal of Medical Genetics 2011;48:73-87.
2. McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet.1994 Dec;8(4):345-51.
3. Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet.1996 Jun;13(2):189-95.
4.Donald J, Pyeritz RE. Hereditary Hemorrhagic Telangiectasia. 2000 Jun 26 [Updated 2017 Feb 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.Available from: https://www.ncbi.nlm.nih.gov/books/NBK1351/
5. Mager JJ1, Overtoom TT, Blauw H, Lammers JW, Westermann CJ. Embolotherapy of pulmonary arteriovenous malformations: long-term results in 112 patients. J Vasc Interv Radiol.2004 May;15(5):451-6.
6. Cottin, V., Dupuis-Girod, S., Lesca, G., & Cordier, J.-F. Pulmonary Vascular Manifestations of Hereditary Hemorrhagic Telangiectasia (Rendu-Osler Disease). Respiration 2007; 74(4), 36178.

