INTRODUÇÃO
A Imunodeficiência Comum Variável (ICV), enquanto imunodeficiência primária, tem uma apresentação heterogénea caracterizada por um aumento da suscetibilidade a infeções, resultante da incapacidade em produzir anticorpos, com défice de Imunoglobulina (Ig) G, IgM ou IgA.1-4Os doentes com ICV podem apresentar uma vasta gama de manifestações clínicas, nomeadamente, infeções bacterianas recorrentes, doenças autoimunes, doença pulmonar intersticial, enteropatia e doenças linfoproliferativas, sendo odiagnóstico geralmente atrasado pela difícil compreensão do distúrbio subjacente.1,2
CASO CLÍNICO
Doente do género masculino, 50 anos de idade, leucodermoide, cantoneiro de profissão, com antecedentes pessoais de alcoolismo, tabagismo (36 UMA) e infeções respiratórias de repetição. Recorreu ao Serviço de Urgência por tosse com expetoração mucopurulenta, ocasionalmente hemoptóica, noção de febre, não mensurada e perda ponderal, não quantificada. Ao exame objetivo salientava-se doente emagrecido, descorado e desidratado, apirético, auscultação pulmonar com roncos bilaterais. A avaliação analítica revelou aumento dos parâmetros inflamatórios de fase aguda leucocitose (29,3x10^3/µL) com neutrofilia absoluta de 27000 neutrófilos/uL, PCR 9.2 mg/dl. Radiografia de tórax com condensação para-hilar esquerda e esboço de imagem espiculada. Assumida pneumonia adquirida na comunidade, iniciou Amoxicilina/Ácido Clavulânico e Azitromicina, tendo ficado internado no Serviço de Medicina Interna. Realizou tomografia computorizada do tórax (TC-Tórax), com evidência de múltiplas bronquiectasias e consolidação no lobo pulmonar inferior esquerdo, associado a uma imagem nodular com 35x25mm (Fig. 1). Presença de adenomegálias mediastínicas, mesentéricas e sub-carinais. Em contexto de suspeita de neoplasia pulmonar, realizou broncofibroscopia, com presença de secreções mucopurulentas abundantes, sem alterações na árvore brônquica. Lavado bronco-alveolar com microbiologia e citologia negativa para células neoplásicas. Após resolução do quadro infecioso, realizou nova TC-Tórax de reavaliação da imagem nodular pulmonar, tendo-se observado resolução da mesma. Em contexto de infeções respiratórias de repetição, com necessidade de múltiplos internamentos nos últimos anos, realizou eletroforese de proteínas séricas com hipogamaglobulinemia grave (Fig. 2). Do estudo imunológico com diminuição marcada de todos os isótopos das IGs, IgA < 27mg/dL; IgM < 17mg/dL; IgG < 30mg/dL; IgE total < 2mg/dL; IgD < 13mg/dL. Imunofenotipagem com Inversão relação CD4/CD8+; linfopenia B acentuada (inferior a 2%); diminuição células B switched e não switched. Serologias HIV 1 e 2 negativas. Imunofixação no soro e urina negativas. Estudo molecular para agamaglobulinemia (BTK) não detetou variantes patogénicas nas regiões analisadas BTK, incluindo transições exão-intrão. Realizou medulograma sem evidência de alterações. Estudo auto-imune negativo. IGRA negativo. Atc anti-Strepto. 0.6mg/L e Anti-toxóide tetânico 0.03IU/mL, após vacinas (ausência de imunidade). Admitido neste contexto, imunodeficiência comum variável, tendo o doente iniciado terapia com Imunoglobulina Humana endovenosa. O doente iniciou tratamento com imunoglobulina humana endovenosa na dose de 600mg/kg (36g para 60Kg) de 4 em 4 semanas. Passados 7 meses sem intercorrências infeciosas.
DISCUSSÃO
A ICV, enquanto imunodeficiência primária, caracteriza-se pela presença de hipogamaglobulinemia, definida de acordo com o intervalo de referência ajustado à idade. O nível de IgG deve ser repetidamente baixo em pelo menos 2 medições com intervalo de pelo menos 3 semanas, podendo ser dispensado um segundo doseamento, se o nível for muito baixo (<100-300 mg/dL, ajustado à idade). O nível de IgA ou IgM deve estar igualmente abaixo dos níveis de referência, ajustados à idade. Certos autores incluem na definição de ICV o défice de IgA. Todos os pacientes com um nível de IgG acima de 100mg/dL devem ser estudados para respostas a antígenos dependentes (TD) e independentes (TI) de T, devendo ser demonstrável uma resposta a pelo menos 1 tipo de antígeno (TD ou TI). Devem ser sempre excluídas outras causas de hipogamaglobulinemia.2 Apesar dos estudos recentes, para explorar a base genética da ICV, ainda não foi possível identificar as mutações genéticas responsáveis pelas manifestações clínicas da ICV.3 Atualmente os estudos genéticos para identificar formas monogênicas ou polimorfismos modificadores da doença não são necessários para o diagnóstico.2,3 As infeções mais comuns na ICV envolvem o sistema respiratório e gastrointestinal. Estas infeções contribuem significativamente para a morbi-mortalidade destes doentes.1,2,4,5 A maioria das infeções são devidas a bactérias, nomeadamente a Streptococcus spp., Haemophilus spp., Moraxella catharralis, Neisseria meningitides e Staphylococcus spp. ou vírus como o Rinovírus, o Herpes zoster e o Mycoplasma spp.5,6 Infeções oportunistas, como Pneumocystis jirovecii e Citomegalovírus, apesar de não serem características, podem ocorrer, principalmente em doentes com défice de células T CD4+.7 O tratamento da ICV passa pela administração de Imunoglobulina Humana por via intravenosa ou subcutânea. A dose ideal de IgG necessária para cada paciente é desconhecida, no entanto a maioria das diretrizes sugere uma dose inicial de 0,4 a 0,5g/kg/mês para imunoglobulina intravenosa e 0,4 a 0,6g/kg/mês para subcutânea. Em caso de enteropatia ou esplenomegalia, existem autores que admitem doses mais elevadas (0,6-0,8g/kg/mês). Os efeitos adversos tendem a estar associados a alterações no próprio produto, taxas de infusão rápida, e em pacientes previamente não tratados ou com tempo significativo entre infusões (> 6 semanas), poderão surgir, mais frequentemente, sintomas como cefaleias, náuseas e vômitos, reações urticariformes, calafrios, mialgias, artralgias, dor abdominal ou lombar. Estes sintomas geralmente cedem à terapêutica anti-histamínica ou anti-inflamatória não esteroide, bem como com à redução da taxa de infusão. Poderão ainda surgir reações tardias, até às 72h, tais como fadiga, cefaleias ou mialgias.2,6 CONCLUSÃO
O diagnóstico da ICV, enquanto diagnóstico de exclusão, apresenta-se como um verdadeiro desafio. A complexidade do diagnóstico pode atrasar o tratamento e piorar o prognóstico. A terapia de reposição com Imunoglobulina Humana é o tratamento de escolha, mas nem sempre impede distúrbios autoimunes ou neoplásicos. A vigilância rigorosa destes doentes é fundamental por forma a evitar complicações infeciosas e não infeciosas que possam ser fatais.
Figura I
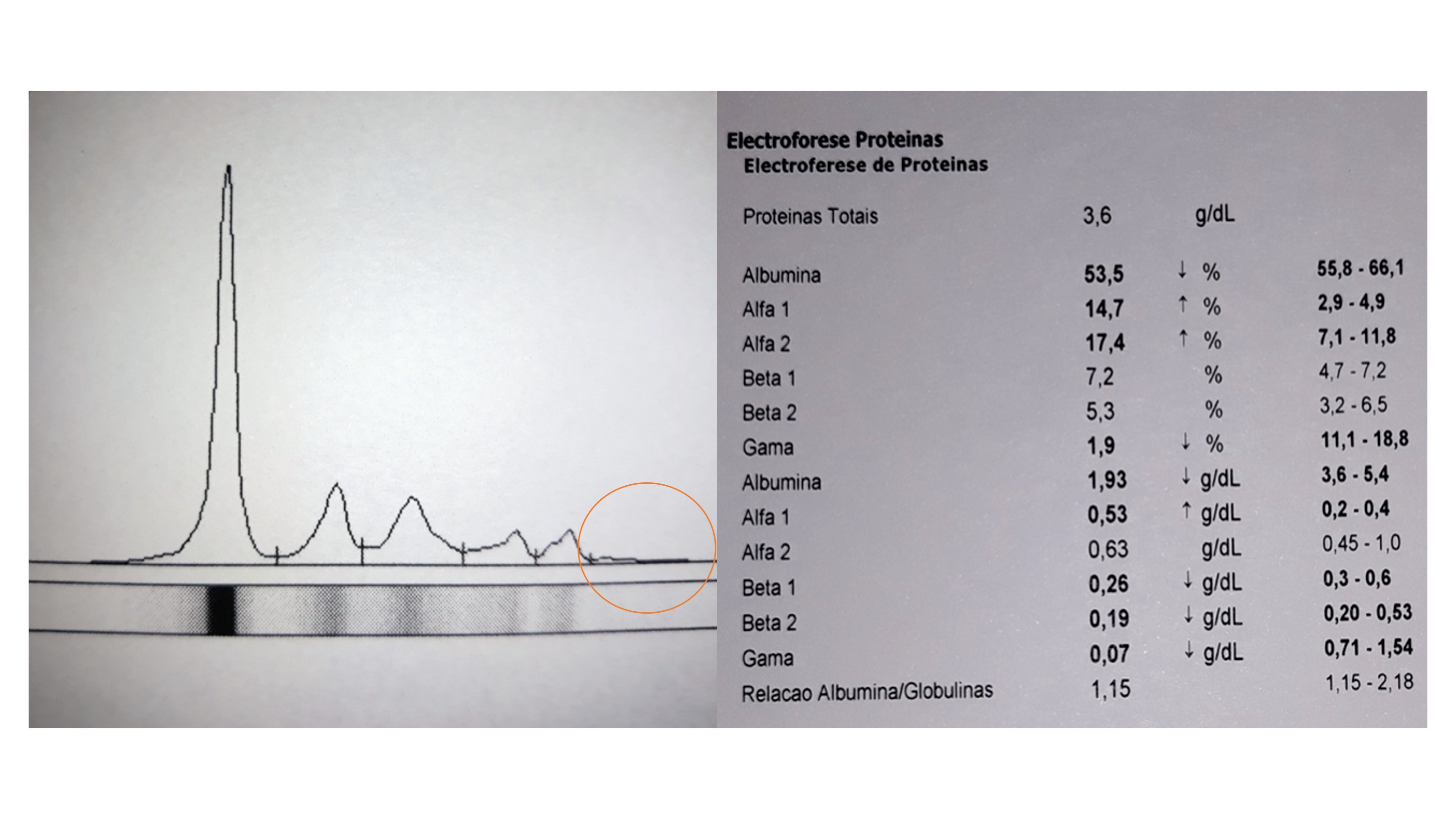
Eletroforese de proteínas séricas com hipogamaglobulinemia grave.
Figura II
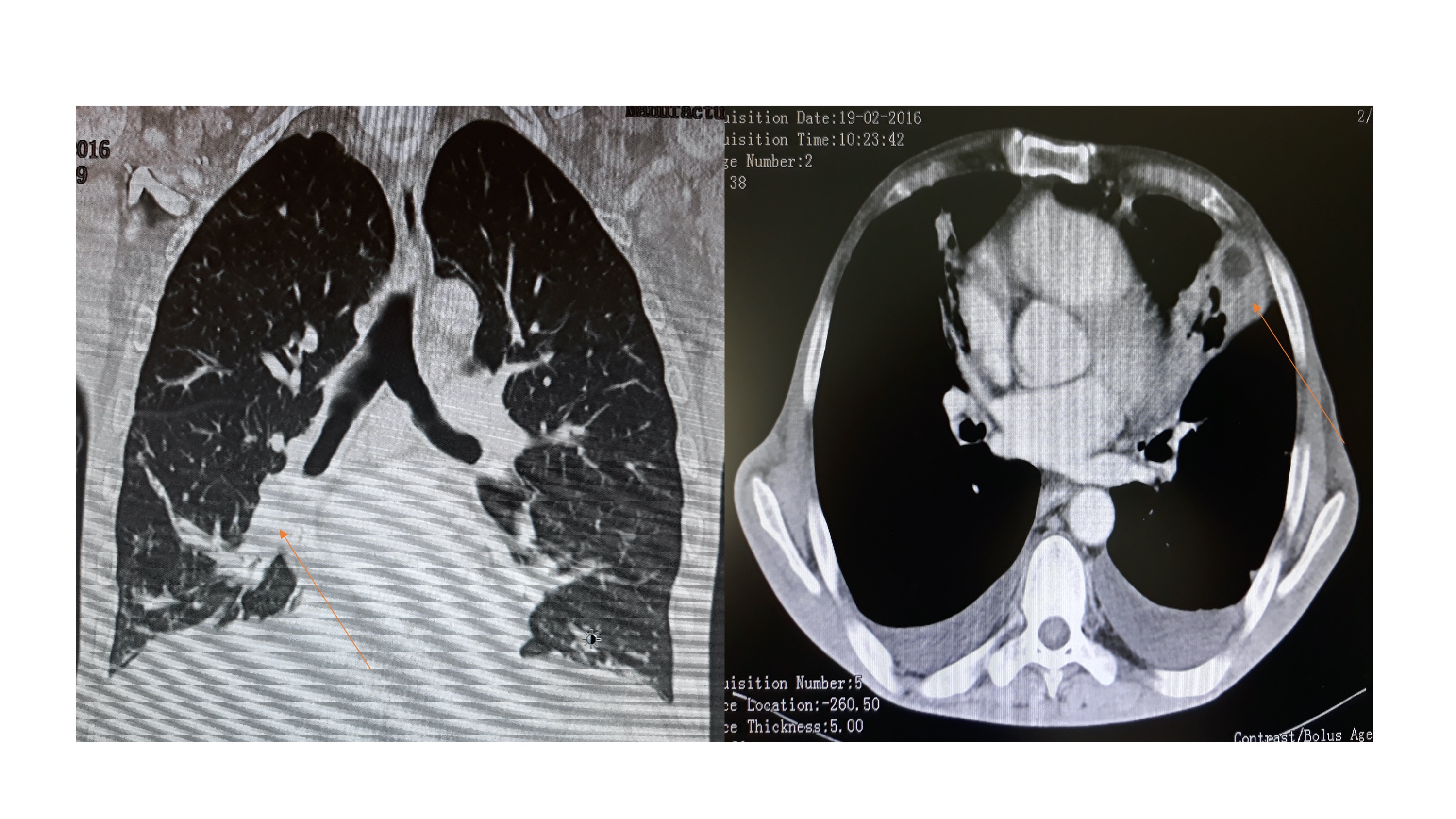
TC-Tórax: Lobo inferior esquerdo com consolidação pulmonar e imagem nodular com 35x25mm. Presença de adenomegálias mediastínicas, mesentéricas e sub-carinais. Múltiplas bronquiectasias bilaterais.
BIBLIOGRAFIA
1.Aghamohammadi A, Allahverdi A, Abolhassani H, Moazzami K, Alizadeh H, Gharagozlou M, et al.Comparison of pulmonary diseases in common variable immunodeficiency and X-linked agammaglobulinaemia. Respirology. 2010;15(2):289-95.
2.Yazdani R , Habibi S , Sharifi L , Azizi G , Abolhassani H , Olbrich P , Aghamohammadi.Common Variable Immunodeficiency: Epidemiology, Pathogenesis, Clinical manifestations, Diagnosis, Classification and Management. J Investig Allergol Clin Immunol 2020 Vol. 30.
3.Warnatz K, Voll RE. Pathogenesis of autoimmunity in common variable immunodeficiency. Front Immunol 2012;3:210.
4.Patuzzo G, Barbieri A, Tinazzi E, Veneri D, Argentino G, Moretta F, et al.Autoimmunity and infection in common variable immunodeficiency (CVID). Autoimmun Rev. 2016;15(9):877
5.Yazdani R, Abolhassani H, Asgardoon M, Shaghaghi M, Modaresi M, Azizi G, et al.Infectious and Noninfectious Pulmonary Complications in Patients With Primary Immunodeficiency Disorders. J Investig Allergol Clin Immunol. 2017;27(4):213-24.
6.Quinti I, Soresina A, Guerra A, Rondelli R, Spadaro G, Agostini C, et al.Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol. 2011;31(3):315-22.
7.Hampson FA, Chandra A, Screaton NJ, Condliffe A, Kumararatne DS, Exley AR, et al. Respiratory disease in common variable immunodeficiency and other primary immunodeficiency disorders. Clin Radiol. 2012;67(6):587-95.

