INTRODUCTION:
Atypical Hemolytic Uremic Syndrome (aHUS) is a very rare and chronic disease originated by systemic thrombotic microangiopathy (TMa) and defined by non-immune hemolytic anemia, thrombocytopenia and acute renal failure.1aHUS has a poor prognosis essentially due to kidney failure, frequently resulting in long periods of RRT (Renal Replacement Therapy) and kidney transplant. In aHUS, inherited genetic variants or acquired autoantibodies lead to an uncontrolled complement dysregulation, which causes microvascular lesion, essentially mediated by c5 activation.2 Complement-mediated damage to endothelial cells lacking cell surface complement inhibitors, results in endothelial swelling, expression of pro-inflammatory cytokines and recruitment of white blood cells and platelets, leading to a pro-thrombotic state.1Eculizumab inhibits the c5 activation, quickly ceasing the aHUS mechanism. This leads to a sustained remission, with the recovery of acute renal failure, if promptly recognized and treated.3There is a particular risk of TMa complications for aHUS patients following exposure to conditions that activate complement or damage the endothelium, including: systemic infections, malignant hypertension, drug exposure, autoimmune disorders or pregnancy.4Elevated maternal complement activation is reported during normal pregnancy.5Complement activation during gestation or the post-partum period may be ascribed to several mechanisms, including immunological incompatibility between the mother and placenta, inflammation, maternalfetal hemorrhage and infections.6 All these conditions may be comorbid with aHUS or unmask a previously undiagnosed case. If the TMa does not cease once the complement activating condition has resolved, clinical suspicion for aHUS should be maintained.7Seven percent of total aHUS and up to 20% aHUS cases in adult females are associated with pregnancy and most of them occurred post-partum.6,8-11This case is an example of how a fast diagnose and a proper prompt treatment made a difference in the prognosis of such a rare and morbid disease.
CASE DESCRIPTION:
31 years old, previously healthy, Caucasian woman, working has a nurse, was submitted to an elective cesarean due to an Arnold-Chiari type 1 malformation, at 38 weeks of pregnancy time. Cesarean was complicated by uterine hemorrhage, with hemodynamic instability, so a total hysterectomy had to be performed. It was her first pregnancy. At the immediate postoperative period, the patient developed acute renal failure (needing RRT), high blood pressure (BP: 157/84mmHg, with normal fundoscopic examination), thrombocytopenia (96.000 x10^9/L platelets) and anemia (Hb 7,0g/dL) with <1% of schistocytes in the blood smear. No other clinical changes were present, fever was never noted and the neurologic exam was always normal. On the following day, anemia persisted in spite of blood transfusion (Hb 6,0 g/dL) and hemolytic non-immune criteria emerged (LDH 434U/L, Total bilirubin: 3.0mg/dL, Haptoglobin < 10mg/dL, Coombs negative), with progressive raise of schistocytes to 4% and a minimum thrombocytopenia of 26.000 x10^9/L. Anuria was noted, with maximum Creatinine value 4,42 mg/dL and Urea 221 mg/dL, and the urinary analysis presented proteinuria in a non-nephrotic range (0.8-2 g/L) and hematuria.
The patient had no extra-renal manifestations, such as neurologic symptoms. At this time, suspecting of a thrombotic microangiopathy, without an established etiology, plasmapheresis every two days and 1g of methylprednisolone per day for 5 days followed by 1mg/Kg/day of prednisolone were started, with very slight improvement. However, the patient remained with renal failure under RRT and persisting hemolytic anemia and thrombocytopenia.
To clarify the etiology of this thrombotic microangiopathy, numerous biochemical, immunological and genetic tests were performed (see Table 1. for further detail).
Considering the diagnosis of TMa, the following hypothesis appeared: thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (Shiga toxin associated or atypical) or secondary TMa.
Secondary TMa:As the patient was a puerperal woman, the most probable hypothesis was a pregnancy related TMa. Although no hypertension and proteinuria were noted during pregnancy, the patient was hypertensive and had proteinuria at the immediate post-partum. Yet, arterial blood pressure was not severely elevated (Systolic BP<160mmHg) and was easily controlled with low dose of nifedipine. Even though preeclampsia could not be totally excluded, the benign course of blood pressure values made us consider that preeclampsia would not likely be the cause for the TMa etiology. HELLP syndrome was not a hypothesis once liver enzymes were normal.
Autoimmune disease, such as primary or secondary Anti-Phospholipid Syndrome (APS) or Systemic Lupus Erythematous (SLE), were excluded considering that: Lupus anticoagulant, anti-cardiolipin, anti-glycoprotein, ANA and anti-dsDNA antibodies were negative; malignant hypertension was not present; infections like HIV or Hepatitis C were also excluded with negative serology. Puerperal sepsis was also excluded by maintained apyrexia, absence of elevated acute phase reagents, no clinical signs or symptoms of infection, and negative blood and urine cultures. Neoplasms were unlikely since the patient had no symptoms supporting that option. Drug associated TMa was not considered because the patient was not taking any suspicious drug (e.g. quinine, calcineurin inhibitors, oxymorphone) before or after delivery.
Thrombotic Thrombocytopenic Purpura:Although neurologic manifestations were not present and renal failure was the major organ manifestation, TTP had to be excluded. So, ADAMTS13 was essential to the differential diagnosis and the results were not concordant with this hypothesis.
Typical Hemolytic Uremic Syndrome:Even though the patient lacked gastrointestinal symptoms, Shiga toxin was searched and negative.
Atypical Hemolytic Uremic Syndrome:The genetic study showed homozygotic deletion of the CFHR1-CFHR3 gene; this deletion is described as being associated with the development of anti-complement H factor antibody, which is confirmed in this case by high values (30,41 UA/mL).12
Hence, immediately after the diagnosis confirmation and exclusion of other possible causes, eculizumab was started on day 15 from disease presentation (900mg intravenous once a week for four weeks, followed by 1200mg every fifteen days for an undetermined time). This treatment allowed progressive improvement of the renal function, anemia and thrombocytopenia. Meanwhile, anti-pneumococci, anti-meningococci and anti-Hemophilus vaccines were given.
After starting eculizumab, RRT was no longer needed, renal function recovered slowly and the patient was discharged at the 25th day, during the induction phase of the treatment (two weeks of treatment with eculizumab 900mg weekly). At discharge, the patient had the following laboratory test results: Hb 8.7g/dL, platelets 141.000x10^9/L, Urea 89mg/dL, Creatinine 1.75mg/dL, albuminuria 24h 1828mg/24h, GFR (measured) 44ml/min/1.73m2.
During follow-up, recovery was documented (see table 2) and the patient kept treatment with eculizumab 1200mg every fifteen days and antibiotic prophylaxis against capsulated agents with amoxicillin 500mg bid.
DISCUSSION:
aHUS is a very rare and life-threatening disease caused by inherited defects of the alternative pathway of complement, characterized by microangiopathic hemolytic anemia, thrombocytopenia and acute kidney injury. It may occasionally be triggered by pregnancy (P-aHUS), mostly in postpartum period.8
Our case was diagnosed at post-partum of the first pregnancy, as appears to be frequent in the case series bellow (table 3).8-11Delivery was via C-section, which is described by Huerta as a trigger factor in 81% of the cases.9
There are only few published cases in which pregnancy ran without any complication, with first symptoms appearing from hours to days after delivery, two also after C-section.13
Considering a puerperal woman with anemia, thrombocytopenia and renal failure, we suspected of TMa. Our patient had no clinical symptoms supporting eclampsia or HELLP syndrome (no seizures or elevated liver enzymes) and had no auto-antibodies for APS and SLE. HIV and HCV serologies were negative, as well as stool cultures for Shiga toxin-producing E. coli or Shiga-like. Moreover, the patient presented no signs of sepsis, ADAMTS13 activity was not reduced or suppressed and anti-ADAMTS13 antibodies were negative. Considering these laboratory tests, we excluded thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (Shiga toxin associated) and most secondary TMa causes. Although we kept preeclampsia as a possible cause, the clinical course made it a less likely diagnosis.
Assuming the aHUS as the most probable etiology, genetic study was performed, finding an homozygotic deletion of the CFHR1-CFHR3 gene. This is described as polymorphic in European population (approximately 2%). However, if homozygotic, it is considered a risk factor for aHUS. Different studies showed association as cumulative risk factor for aHUS in the presence of precipitating factors, but it is controversial if it is a causeper seof TMa.14,15
Immediately after aHUS strong suspicion, the patient started eculizumab 900mg intravenous once a week for four weeks followed by 1200mg every fifteen days. Eculizumab is a humanized monoclonal antibody that binds with high affinity to the human C5 complement protein and blocks the generation of C5a and C5b-9.3It is approved for the treatment of paroxysmal nocturnal hemoglobinuria, aHUS, myastenia gravis and, most recently, neuromyelitis optica spectrum disorder.
Eculizumab should be started as soon as possible, since it strongly impacts prognosis, especially in the recovery of renal function. However, case rarity, delay in laboratory results and drug availability postponed its initiation.
The patient evolved without any treatment complication, with immediate improvement and total renal function recovery in the following six months. After 15 months the patient is still under treatment for undetermined time, with no disease relapse until present time.
As far as we know, this is the first case of aHUS pregnancy-related reported in the Portuguese population, submitted to eculizumab treatment, being free from renal replacement therapy with total renal function recovery.
Figura I
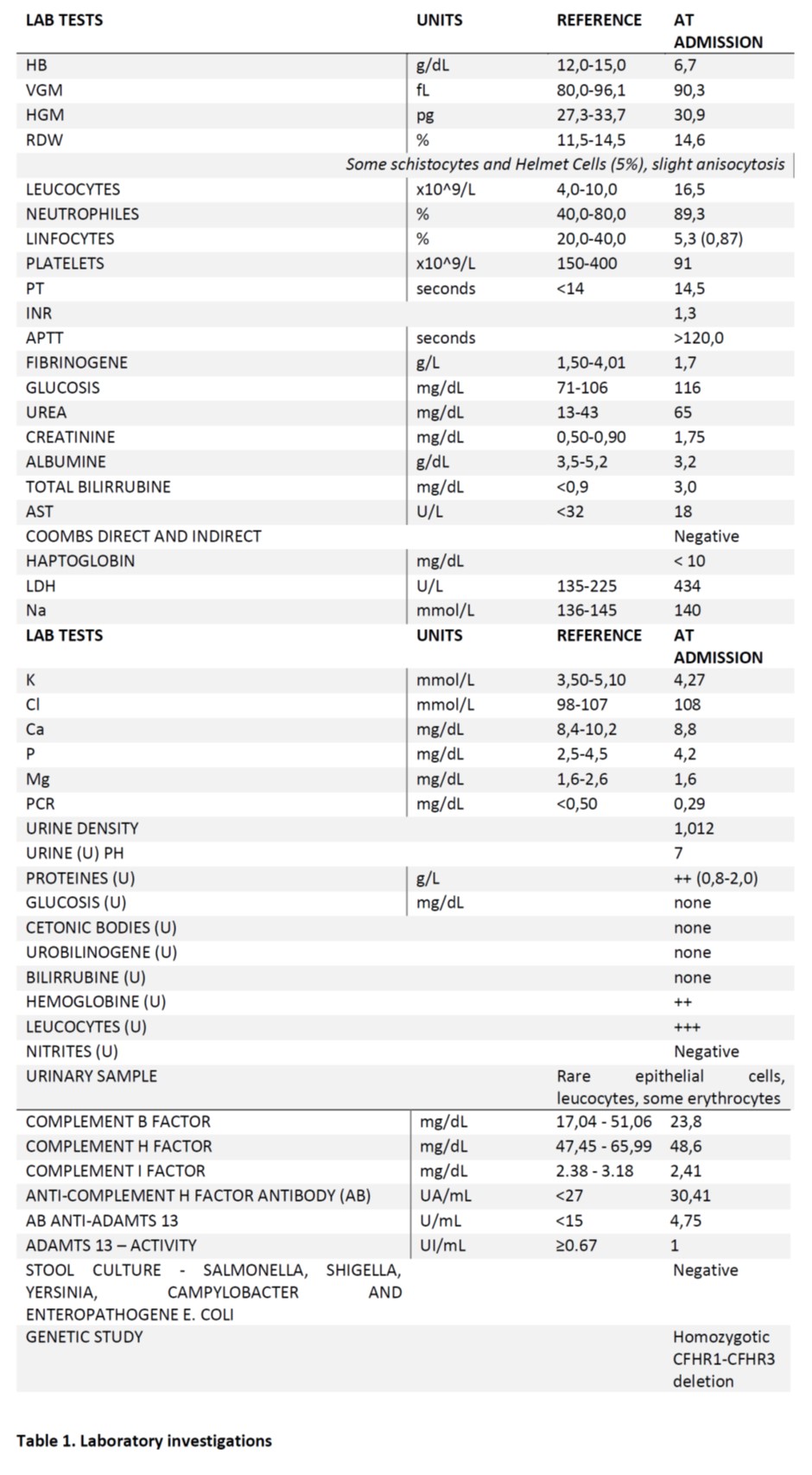
Table 1. Laboratory investigations
Figura II
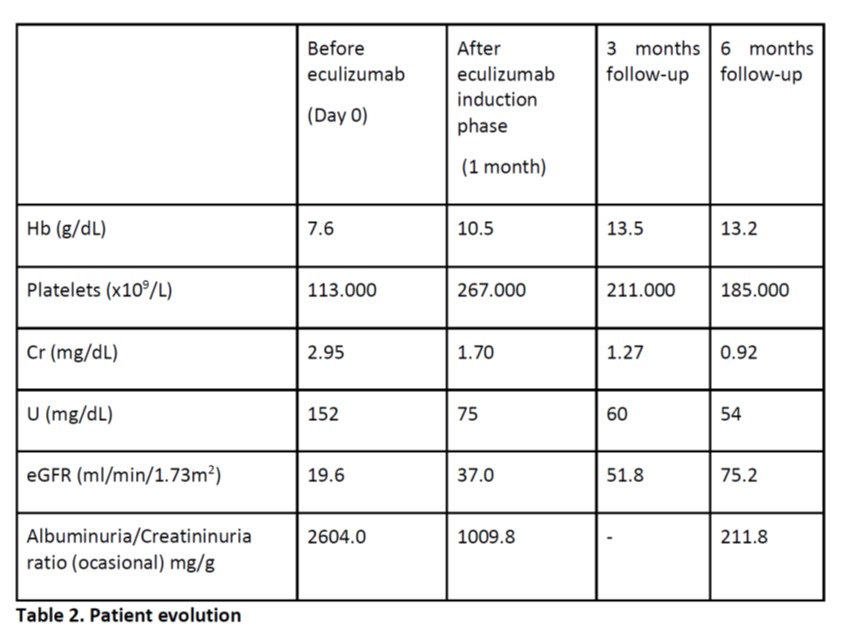
Table 2. Patient evolution
Figura III
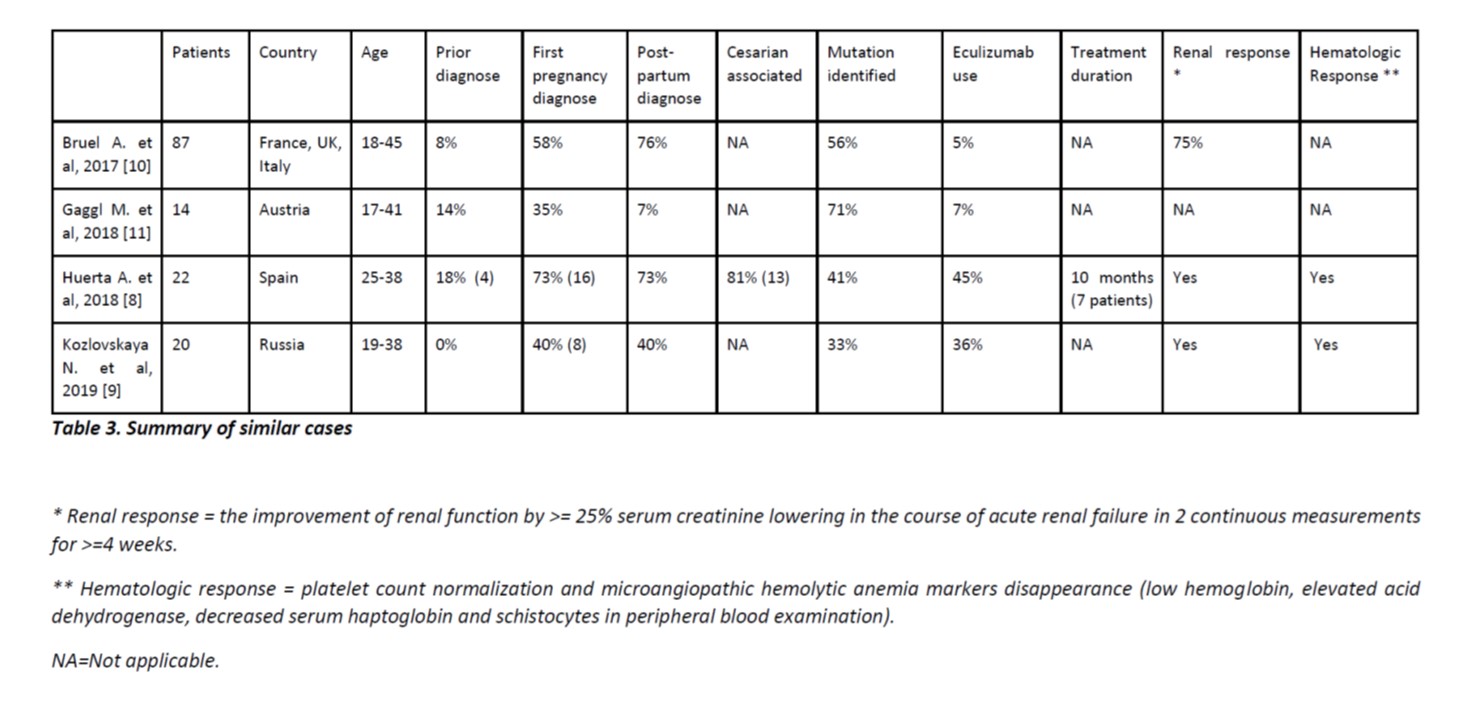
Table 3. Summary of similar cases
BIBLIOGRAFIA
1. Noris M, Remuzzi G. Atypical HemolyticUremic Syndrome. N Engl J Med. 2009 Oct 22;361(17):167687. https://doi.org/10.1056/nejmra0902814
2. Carroll M V, Sim RB. Complement in health and disease. Adv Drug Deliv Rev. 2011;63(12):96575. https://doi.org/10.1016/j.addr.2011.06.005
3. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal Complement Inhibitor Eculizumab in Atypical HemolyticUremic Syndrome. N Engl J Med. 2013 Jun 5;368(23):216981. https://doi.org/10.1056/nejmoa1208981
4. Asif A, Nayer A, Haas CS. Atypical hemolytic uremic syndrome in the setting of complement-amplifying conditions: case reports and a review of the evidence for treatment with eculizumab. J Nephrol. 2017;30(3):34762. https://doi.org/10.1007/s40620-016-0357-7
5. Regal JF, Gilbert JS, Burwick RM. The complement system and adverse pregnancy outcomes. Mol Immunol. 2015;67(1):5670. https://doi.org/10.1016/j.molimm.2015.02.030
6. Fakhouri F, Roumenina L, Provot F, Sallée M, Caillard S, Couzi L, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010/03/04. 2010 May;21(5):85967. https://doi.org/10.1681/ASN.2009070706
7. Laurence J, Haller H, Mannucci PM, Nangaku M, Praga M, Rodriguez de Cordoba S. Atypical hemolytic uremic syndrome (aHUS): essential aspects of an accurate diagnosis. Clin Adv Hematol Oncol. 2016;14(Suppl 11):215.
8. Huerta A, Arjona E, Portoles J, Lopez-Sanchez P, Rabasco C, Espinosa M, et al. A retrospective study of pregnancy-associated atypical hemolytic uremic syndrome. Kidney Int. 2018 Feb;93(2):4509. https://doi.org/10.1016/j.kint.2017.06.022
9. Kozlovskaya NL, Korotchaeva Y V, Bobrova LA. Adverse outcomes in obstetric-atypical haemolytic uraemic syndrome: a case series analysis. J Matern Fetal Neonatal Med. 2018 Apr;17.https://doi.org/10.1080/14767058.2018.1450381
10. Bruel A, Kavanagh D, Noris M, Delmas Y, Wong EKS, Bresin E, et al. Hemolytic Uremic Syndrome in Pregnancy and Postpartum. Clin J Am Soc Nephrol. 2017/06/08. 2017 Aug 7;12(8):123747. https://doi.org/10.2215/CJN.00280117
11. Gaggl M, Aigner C, Csuka D, Szilágyi Á, Prohászka Z, Kain R, et al. Maternal and fetal outcomes of pregnancies in women with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2018;29(3):10209. https://doi.org/10.1681/ASN.2016090995
12. Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement factor H related proteins (CFHRs). Mol Immunol. 2013;56(3):17080. https://doi.org/10.1016/j.molimm.2013.06.001
13. Cañigral C, Moscardó F, Castro C, Pajares A, Lancharro A, Solves P, et al. Eculizumab for the treatment of pregnancy-related atypical hemolytic uremic syndrome. Ann Hematol. 2014;93(8):14212. https://doi.org/10.1007/s00277-013-1970-3
14. Abarrategui-garrido C, Lo M. Characterization of complement factor H related ( CFHR ) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. 2019;114(19):426172. https://doi.org/10.1182/blood-2009-05-223834
15. Richter H, Misselwitz J, Hoppe B, Zipfel PF, Edey M, Heinen S, et al. Deletion of Complement Factor H Related Genes CFHR1 and CFHR3 Is Associated with Atypical Hemolytic Uremic Syndrome. 2007;3(3). https://doi.org/10.1371/journal.pgen.0030041

