Introdução
A amiloidose de cadeias leves (AL), historicamente referida como amiloidose primária, é uma doença sistémica caraterizada pela deposição progressiva de imunoglobulina de cadeias leves produzida por um clone de células plasmáticas levando a depósitos de fibrilhas de amiloide e disfunção de órgão. A amiloidose primária está raramente associada a doenças linfoproliferativas de células B, cujos aspectos patológicos são mais característicos de macroglobulinémia deWaldenström, leucemia linfocítica crónica (LLC), ou linfoma linfoplasmocítico. 1,2,3. Alguns aspectos clínicos dos doentes com amiloidose AL associada com doenças linfoproliferativas podem distingui-los dos doentes com amiloidose AL associada mais tipicamente a discrasias das células plasmáticas como Mieloma múltiplo (MM), designadamente os doentes são em média mais idosos, mais frequentemente têm doença amiloidotica multissistémica e mais provavelmente têm gamapatia monoclonal IgM. 2
Caso clínico
Mulher de 78 anos, caucasiana, reformada, admitida no Serviço de Urgência por quadro com cerca de 6 meses de evolução de perda ponderal, anorexia e fadiga; negava febre, queixas cardiorrespiratórias, gastrointestinais ou urinárias, bem como edema e parestesias dos membros. Negava história heredofamiliar de hemopatias malignas. Estava medicada com furosemido, omeprazol, AAS, sinvastatina, bromazepam e temazepam.
Tinha antecedentes de hipertensão arterial, dislipidemia e AVC isquémico em 2017; era referida cardiopatia isquémica,da qual não se encontrou ecocardiograma prévio. À entrada a doente apresentava-se consciente e orientada, emagrecida e eupneica, PA 108/57 mmHg, FC 84 bpm, TT 37.4º; mucosas descoradas, ausência de macroglossia e adenopatias cervicais milimétricas, de consistência elástica, não aderentes.A auscultação cardiopulmonar não revelava alterações e o abdómen estava ligeiramente distendido com hepatomegalia moderada, sem esplenomegalia. No exame neurológico sumário salientava-se hemiparésia esquerda de predomínio braquial e ausência de alterações da sensibilidade.
Da avaliação laboratorial destacava-se Hb 7.5 g/dL, hematócrito 24%, V.G.M. 80.8 fL, leucócitos 9.8x10^9/L, neutrófilos 63,9%, linfócitos 24%, plaquetas 182x10^9/L, velocidade de sedimentação 99 mm na 1ª hora, cálcio sérico 9.2mg/dL, creatinina 2.53 mg/dL (2,2 mg/dL cerca de 3 meses antes), ureia 94 mg/dL, proteínas totais 7.36 g/dL e sedimento urinário com hematoproteinúria. As provas de função hepática e da coagulação não mostravam alterações. Teve ferritina de 200 ng/mL; as serologias virais, o estudo da autoimunidade, incluíndo complemento e crioglobulinas foram negativos. As hormonas tiroideias e os doseamentos da vitamina B12 e ácido fólico estavam normais. O ECG mostrava ritmo sinusal e padrão de BRE e a radiografia do tórax não apresentava infiltrados pulmonares.
Na investigação subsequente tinha proteinúria de 1824,83 mg nas 24 horas; a eletroforese sérica mostrou pico monoclonal de IgM 3417,5 mg/dL e a imunofixação sérica revelou proteinémia de cadeias leves livres (Bence Jones), com hipogamaglobulinémia envolvendo IgG e IgA. O doseamento sérico e urinário das cadeias livres revelou marcado aumento das cadeias leves livres lambda respetivamente de 416,53 mg/L (N: 5,71- 26,3) e 194,78 mg/L (N: 0,8-10,10), com relação cadeias livresκ /λde 0,191 (N:0,26-1,65), indicando sobreprodução das cadeias leves lambda. Teve beta 2 microglobulina de 35,26 mg/L.
A radiografia do esqueleto não revelou lesões osteolíticas. Fez ainda ressonância magnética da coluna cervical e dorsal que sugeriu múltiplos hemangiomas vertebrais. A endoscopia digestiva alta revelou hiperémia do antro.
A ecografia renal mostrou rins de contornos regulares e dimensões normais com espessura parenquimatosa mantida, observando-se, contudo, aumento da ecogenicidade do parênquima que condicionava redução da diferenciação parenquimo-sinusal; bilateralmente sem ectasia das cavidades excretoras e sem sinais seguros de litíase ou lesões expansivas.
A tomografia computorizada de pescoço, tórax, abdómen e pélvis (sem administração de contraste endovenoso pela insuficiência renal) identificou algumas adenomegalias cervicais, axilares e inguinais infra-centimétricas, ascite de pequeno volume e fígado moderadamente aumentado sem lesões nodulares e rins no limite inferior da normalidade, com espessura do parênquima mantida, sem evidência de litíase ou dilatação do sistema excretor.
Perante o quadro de anemia e de insuficiência renal crónica agravada em associação a um pico monoclonal de cadeias leves livres lambda procedeu-se a mielograma e BO; o primeiro revelou série linfóide aumentada constituída predominantemente por pequenos linfócitos com morfologia aparentemente madura, megacariócitos em número normal e raros plasmócitos. A biópsia osteomedular mostrou plasmocitose madura intersticial (cerca de 15%) sem evidência imuno-histoquímica de monoclonalidade e infiltração paratrabecular e intersticial por pequenos linfócitos maduros que expressavam imuno-histoquímicamente positividade para CD79a, CD20 e CD38, sendo negativa para CD23, aspectos sugestivos de linfoma de pequenas células B com diferenciação plasmocitica (macroglobulinemia deWaldenström) (Imagem 1a e 1b).A pesquisa histoquímica da substância amiloide foi negativa (coloração Vermelho do Congo)
Para esclarecimento da etiologia da insuficiência renal, a Hematologia recomendou biópsia renal, a qual revelou ao nível da parede dos vasos, no interstício e em mais de 50% glomérulos, depósito de material amorfo e hialino que corava a vermelho com a coloração histoquímica de Vermelho do Congo e tinha birrefringência verde maçã à luz polarizada, compatível com amiloidose.(Imagem 2a e 2b) A pesquisa imuno-histoquímica de amiloide AA foi negativa. O estudo pela imunofluorescência revelou expressão intensa de cadeias leves lambda no mesângio e na parede capilar dos novelos glomerulas, no interstício e na espessura dos vasos (zonas de depósito da substância amilóide)- compatível com o diagnóstico de amiloidose AL.
A biópsia renal mostrou ainda permearão difusa do interstício e do tecido adiposo periférico por pequenas células redondas e azuis que expressavam de forma intensa e difusa CD20 e CD79a, compatível com linfoma de células B de baixo grau- provável linfoma linfoplasmocítico.(Imagem 3a e 3b)
A doente teve agravamento da função renal atingindo creatinina sérica de 6.11g/dl, pelo que fez indução dialítica transitoriamente com estabilização (Cr plasmática 2,67 mg/dL).
Após o resultado de amiloidose AL na biópsia renal, realizou ecocardiograma transtorácico que mostrou ventrículo esquerdo de dimensões normais, parede ligeiramente hipertrofiada (13-14 mm), com ecogenicidade heterogénea (padrão mosqueado) com fração de ejeção preservada, mas comstrainglobal longitudinal diminuído (-11%), poupando os segmentos apicais, sugestivos de doença infiltrativa do miocárdio. Dilatação ligeira da aurícula esquerda; disfunção diastólica grau I e relação E/e´=18, sugestivo de aumento das pressões de enchimento do VE. Sem dilatação das cavidades direitas ou da veia cava inferior. Sem derrame pericárdico.
A doente registou deterioração progressiva do seu estado geral, não a candidatando a hemodiálise ou quimioterapia, vindo a falecer cerca de 2 meses após o diagnóstico de amiloidose AL.
DISCUSSÃO
A gamapatia monoclonal IgM pode ocorrer em diferentes doenças hematológicas, constituindo um desafio diagnóstico para os clinicos . Por sua vez, a amiloidose AL está tipicamente associada a doenças proliferativas das células plasmáticas, nomeadamente ao mieloma múltiplo ; a sua associação com macroglobulinémia deWaldenströmestá descrita muito raramente em casos isolados ou em pequenas séries. 1,2,4,5
O caso apresentado combinava amiloidose AL na biópsia renal , presença de gamapatia monoclonal IgM (sérica) e infiltração da medula por linfócitos pequenos com padrão de diferenciação plasmocítica e imunofenotipo típico , evocando critérios de diagnóstico de macroglobulinémia deWaldenström 6. Deveria estar presente no diagnóstico diferencial a amiloidose AL IgM, a qual representa 5 a 7 % de todos os doentes com amiloidose AL e que está reportada em cerca de 7,5% dos doentes com macroglobulinémia de Waldenström /linfoma linfoplasmocítico e amiloidose , dos quais cerca de metade têm o duplo diagnóstico de MW e amiloidose.5 Os estudos citogenéticos adicionais na medula óssea poderiam caracterizar melhor a infiltração neoplásica , nomeadamente com a pesquisa da mutação MYD88 , a qual é geralmente positiva na MW , por alguns considerada até patognomónica , mas insuficiente para critério isolado de diagnóstico de MW.6
O Mieloma Múltiplo IgM e a MW são duas doenças hematológicas diferentes com o achado comum de gamapatia monoclonal IgM. O MM IgM é uma entidade rara ,representando menos de 0,5 % de todos os casos de mieloma. . No caso o apresentado a presença de adenopatias e a ausência de hipercalcémia e de lesões liticas não favoreciam o diagnóstico de MM IgM; a mutação MYD88 geralmente ausente no MM IgM tamém aqui ajudaria no diagnóstico diferencial.6
A MW, a par do linfoma linfoplasmocítico e da LLCpartilham marcadores imuno-histoquímicos comuns; no entanto, no caso descrito a ausência de hiperleucocitose e de linfocitose no sangue periférico não sugeria LLC e a presença dum pico monoclonal IgM tão exuberante também não é um aspecto descrito na LLC. (7).O linfoma linfoplasmocítico, por sua vez, não é sinónimo de MW; é um linfoma indolente que frequentemente envolve a medula óssea, sangue periférico e baço e no qual a paraproteína monoclonal IgM está ausente8
A MW é uma doença rara, situando-se a idade média de diagnóstico entre os 63 e os 68 anos, sendo incomum em idades inferiores a 40 anos. Na maioria dos casos as manifestações clínicas são atribuíveis à presença da paraproteína monoclonal IgM em circulação e/ou a infiltração de órgãos e sistemas,embora não haja correlação entre sintomas e o valor da concentração da proteína monoclonal IgM 3.Pode acompanhar-se de sintomas de hiperviscosidade, fenómenos autoimunes ou neuropatias .Os factores associados com o prognóstico da MW e que estratificam a categoria de risco em baixo, intermédio e alto são a idade >65 anos, hemoglobina <11,5 g/dl, plaquetas <100 K/microlitro, B2 microglobulina> 3 mg/dl e concentração da IgM monoclonal >7g/dl 3
Do ponto de vista clínico a doente não apresentava síndrome de hiperviscosidade, nomeadamente queixas de hemorragia nasal ou das gengivas, queixas oculares como visão turva ou perda da visão bem como cefaleias e vertigens, entre outros sintomas, o que ocorre na macroglobulinémia deWaldenström, particularmente quando o componente M da IgM é superior a 5g/dL. 9
Na amiloidose AL qualquer órgão, excepto o SNC, pode ser um local de deposição de amiloide AL; os locais mais comuns de envolvimento incluem rim, fígado, coração, sistema nervoso periférico, sistema musculosquelético e pele. O rim está afectado em 50-80% dos casos e aproximadamente 50% dos doentes têm envolvimento cardíaco, manifestando-se por insuficiência cardíaca rapidamente progressiva; por vezes os doentes têm angina típica por infiltração amiloide de pequenos vasos e síncope postural por disfunção autonómica.10Na doente os achados ecocardiográficos são muito sugestivos de envolvimento cardíaco, embora com repercussão moderada, com disfunção diastólica grau I (alteração do relaxamento), sem fisiologia restritiva, sem dilatação da veia cava inferior e sem derrame pericárdico
O prognóstico dos doentes com linfomas B, nomeadamente a MW, associados a amiloidose AL permanece pouco claro sendo que o envolvimento cardíaco confere um prognóstico significativamente pior.11No caso apresentado, contudo, as manifestações de insuficiência cardíaca eram ligeiras, pelo que o atingimento cardíaco pela amiloidose não parece ter sido o determinante major no desfecho fatal.
A opção terapêutica preferida na MW é a combinação de rituximab com ciclofosfamida e dexametasona, embora mais recentemente têm sido introduzidos, com benefício, novos agentes, como talidomida e bortezomib . No entanto, a terapêutica óptima para os doentes com amiloidose AL e macroglobulinémia deWaldenströmé ainda desconhecida, mas provavelmente deverá consistir de drogas activas contra ambos os componentes linfoide e de células plasmáticas, nomeadamente regime de combinação com rituximab. 3
Apesar do investimento diagnóstico com os exames invasivos referidos, as opções terapêuticas da doente decididas em ambiente multidisciplinar foram, no entanto, muito limitadas pelo seu estado geral , que de forma rápida se deteriorou.
Conclusão
Os autores documentam a associação rara de amiloidose AL e MW, duas entidades por sua vez muito incomuns, numa doente de78anos, com fadiga, anorexia, proteinúria não nefrótica e insuficiência renal. Teve amiloidose renal e cardíaca e infiltração renal e da medula óssea por linfoma linfoplasmocítico a par de paraproteína sérica IgM monoclonal confirmativos de MW. No caso apresentado, e aliás de acordo com a literatura, a idade avançada, o atingimento amiloidótico multissistémico e a gamapatia monoclonal IgM , são aspectos distintivos da associação rara da amiloidose AL com MW , que a diferenciam da associação mais típica da amiloidose AL com mieloma múltiplo.
Figura I
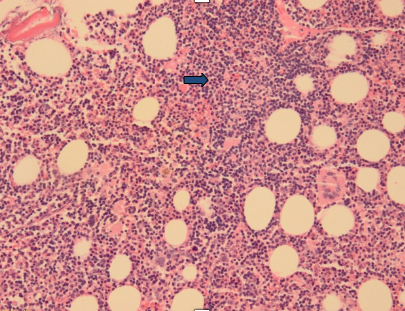
1a- (Hematoxilina e eosina HE- 10x): biópsia osteomedular com espaço intertrabecular com permeação intersticial difusa por uma população de células pequenas, redondas e azúis (seta azul).
Figura I
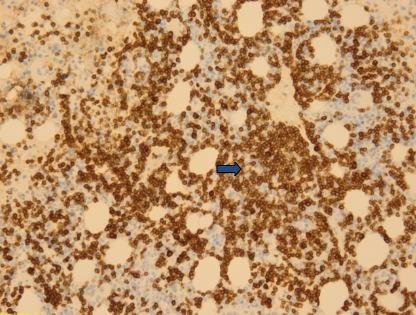
1- (Estudo imunohistoquímico com CD79- 10x): biópsia osteomedular com espaço intertrabecular com permeação intersticial difusa por uma população de células pequenas, redondas e azúis e que expressam CD79 (marcador de linfócitos B) (seta azul).
Figura II
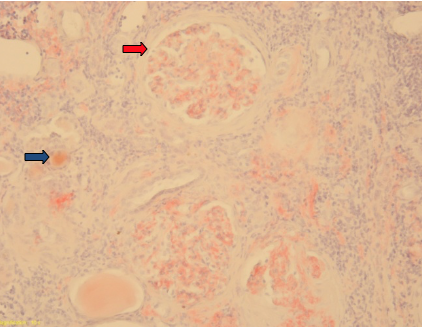
2a- (Coloração histoquímica Vermelho do Congo- x10): região cortical renal com depósito de substância amilóide, evidenciada a vermelho, no novelo glomerular (mesângio e parede capilar) (sete vermelha) e na espessura da parede dos vasos (seta azul).
Figura II
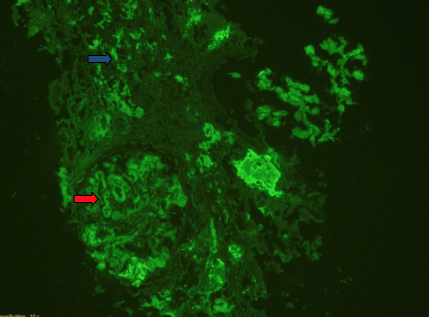
Imagem 2b (Estudo por Imunofluorescência - cadeias leves lambda- x10): expressão de cadeias leves lambda nas áreas de depósito da substância amiloide(mesângio e parede capilar dos glomérulos- sete vermelha) e no interstício (sete azul)- compatível com Amiloidose AL.
Figura III
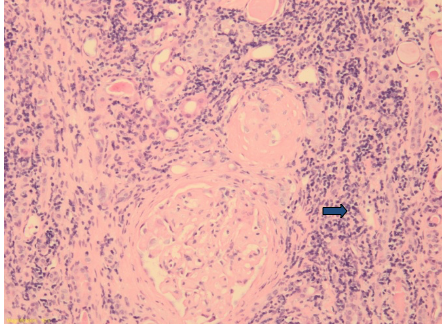
Imagem 3a (Hematoxilina e eosina HE- 10x): região cortical com permeação intersticial difusa por uma população de células pequenas, redondas e azúis (seta azul).
Figura III
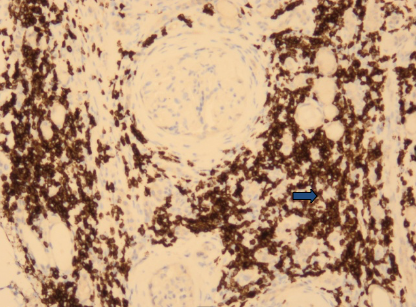
Imagem 3b (Estudo imunohistoquímico com CD79- 10x): região cortical com permeação intersticial difusa por uma população de células pequenas, redondas e azúis e que expressam CD79 (marcador de linfócitos B) (seta azul).
BIBLIOGRAFIA
1. Taxiarchis V. Kourelis, Morie Gertz, Clive Zent, Martha Lacy, Robert Kyle, Prashant Kapooret al. Systemic amyloidosis associated with chronic lymphocytic leukemia/ small lymphocyticlymphoma. American Journal of Hematology 88:375-378, 2013.
2. V. Sanchorawala, E. Blanchard, D.C. Seldin, Carl O Hara, M. Skinner, D.G. Wright. ALAmyloidosis Associated with B- Cell Lymphoproliferative Disorders: Frequency and TreatmentOutcomes. American Journal of Hematology 81: 692-695, 2006.
3. Gertz MA.Waldenstrom macroglobulinemia.. Hematology.2012;17 Suppl 1: S112-6.
4. A. D. Cholen, P. Zhou, Q. Xiao, M. Fleisher, N. Kalakonda, T. Akhurst et al. Systemic ALamyloidosis due to non- Hodgkins lymphoma: an unusual clinopathologic association. BritishJournal of Hematology 124, 309-314, 2004.
5 .Sidana, S., Larson, D.P., Greipp, P.T. et al. IgM AL amyloidosis: delineating disease biology andoutcomes with clinical, genomic and bone marrow morphological features. Leukemia 34, 13731382(2020). https://doi.org/10.1038/s41375-019-0667-6
6. Elba, S., Castellino, A., Soriasio, R. et al. Immunoglobulin M (IgM) multiple myeloma versusWaldenström macroglobulinemia: diagnostic challenges and therapeutic options: two case reports. J MedCase Reports 14, 75 (2020). https://doi.org/10.1186/s13256-020-02380-2 .
7. David Telio, Denis Bailey, Christine Chen, Michael Crump, Donna Reece, Vishal Kukreti. Twodistinct syndromes of lymphoma associated AL amyloidosis: A case series and review ofliterature. American Journal of Hematology 85: 805-808, 2010.
8. Campo E, et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolvingconcepts and practical applications .Blood 2011;117:5019-32
9. Lee Goldman, Andrew I Schafer. Goldman- Cecil Medicine. Philadelphia, PAElsvier/Saunders; 2016. P.1284-1286
10.Douglas P. Zipes, Peter Libby, Robert O. Bonow, Douglas L. Mann, Gordon F Tomaselli.Braunwalds Heart Disease: A Textbook of Cardiovascular Medicine. Philadelphia, PA:Elsevier/Saunders. 2018; p. 1580-160
11.Vaxman I, Gertz M. Recent Advances in Diagnosis, Risk Stratification, and management ofSystemic Light- Chain Amyloidosis. Acta Haematologica 141, 93-106, 2019

