Introdução
A linfangioleiomiomatose (LAM) é uma doença rara, com uma prevalência de aproximadamente 5 por milhão, sistémica e progressiva, que afeta predominantemente mulheres jovens e é caracterizada por destruição quística pulmonar, acumulação de líquido quiloso e tumores abdominais, incluindo angiomiolipomas e linfangioleiomiomas.1-2
Em geral, a LAM ocorre esporadicamente (S-LAM); contudo, em 40% dos casos, afeta mulheres com complexo de esclerose tuberosa (TSC-LAM), síndrome hereditário associado a convulsões, comprometimento cognitivo e formação de tumores em múltiplos órgãos.1-2 As manifestações clínicas mais frequentes são pneumotoraces recorrentes, dispneia progressiva, tosse, dor torácica e hemoptises3-4. Histologicamente, a LAM é caracterizada pela proliferação imatura das células de músculo liso, comummente conhecidas como células LAM.2,5 A proliferação leva à ativação inapropriada da via de sinalização intracelular de mamíferos da rapamicina (mTOR), responsável pelo ciclo de regulação das células.5 Sirolimus (rapamicina), um inibidor da mTOR, é atualmente o tratamento recomendado, melhorando a função pulmonar (FVC e FEV1), desempenho funcional, a qualidade de vida e reduzindo o tamanho dos angiomiolipomas, linfangioleiomiomas e acumulação quilosa.1,5
Reportamos o caso de uma senhora de 40 anos de idade com o diagnóstico de TSC-LAM, cuja primeira manifestação clínica foi um pneumotórax espontâneo e atualmente se encontra a realizar tratamento com sirolimus.
Caso clínico
Doente do género feminino, 40 anos, costureira, ex-fumadora desde há 1 ano (7,6 unidades-maço-ano) com antecedentes de esclerose tuberosa diagnosticada aos 30 anos, angiomiolipomas renais com seguimento em consulta de transplante renal e toma regular de anticoncetivos orais, recorre ao serviço de urgência por quadro clínico com cerca de 1 mês de evolução, caracterizado por dispneia rapidamente progressiva, tosse produtiva de expetoração mucosa e, nos dois dias precedentes ao internamento, toracalgia esquerda de características pleuríticas.
Na avaliação clínica inicial, encontrava-se subfebril, taquicárdica, polipneica com tiragem intercostal, à auscultação pulmonar apresentava diminuição do murmúrio vesicular no hemitórax esquerdo e sibilância difusa. Gasimetricamente revelava insuficiência respiratória parcial e alcalose respiratória (pH 7,48; PCO2: 27mmHg; PO2: 53mmHg) e, analiticamente, apenas ligeira elevação da Proteina-C-Reativa (PCR) de 2,87 mg/dl.
A radiografia de tórax à entrada apresentava padrão intersticial bilateral de predomínio basal e hipertransparência no andar inferior do campo pulmonar esquerdo, compatível com pneumotórax espontâneo (Fig. 1).
A doente, admitida no serviço de Pneumologia, foi submetida a toracostomia com drenagem subaquática, oxigenoterapia e iniciou antibioterapia com levofloxacina, apresentando melhoria clínica e resolução do pneumotórax.
A tomografia computorizada de tórax de alta resolução (TCAR) identificou inúmeras cavidades quísticas aéreas de parede fina e distribuição difusa bilateral (Fig. 2) e a abdomino-pélvica vários angiomiolipomas renais o maior de 18mm de diâmetro. A TC e RMN craneoencefálica revelaram calcificações justa-ventriculares e subependimárias sugestivas de esclerose tuberosa (Fig. 3) O restante estudo analítico revelou hipercolesterolémia e hipertrigliceridémia, sendo medicada com estatina, fenofibrato e dieta hipolipídica. O vascular endotelial growth factor-D (VEGF-D) encontrava-se normal (6pg/mg).
O estudo funcional ventilatório revelou uma síndrome obstrutiva moderada (FEV1: 55,9%. FVC: 80,7% FEV1/FVC:59,5% do valor previsto) com resposta positiva à broncodilatação e uma capacidade de difusão muito diminuída (DLCO:32,6%). Na prova de marcha de 6 minutos, percorreu 429 metros (55% do previsto) apresentando dessaturação até aos 81%.
Os achados encontrados são compatíveis com o diagnóstico de linfangioleiomiomatose com complexo de esclerose tuberosa. A doente iniciou terapêutica broncodilatadora com brometo de glicopirrónio (43 µg), indacaterol (85 µg), corticoterapia inalada (budesonida 400 µg), oxigenoterapia de deambulação 3L/min e terapêutica com Sirolimus 1mg/dia. O doseamento sérico de sirolimus foi realizado às 2 semanas após o início do tratamento e, posteriormente, de 3 em 3 meses, encontrando-se sempre dentro dos valores alvo desejados (1 a 5 µg/L).
Após o início do tratamento como efeitos secundários a registar pequena mucosite oral controlada com corticóoide tópico e diarreia ligeira autolimitada nos primeiros dias de tratamento.
A doente apresentou melhoria progressiva da dispneia, com capacidade para realizar as suas atividades diárias e laborais e o estudo funcional ventilatório, repetido aos 3 e 6 meses após início de terapêutica, apresentou estabilidade da função pulmonar.
Oito meses após o início de tratamento, a doente apresentou novo pneumotórax espontâneo secundário, tendo sido submetida a pleurodesis cirúrgica por video-assisted thoracoscopic surgery (VATS). É, neste momento, candidata para transplantação pulmonar.
Discussão
A LAM é uma doença multissistémica caracterizada por lesões quísticas pulmonares e lesões extrapulmonares que consistem em angiomiolipomas renais e envolvimento linfático4.
E uma doença rara que ocorre quase exclusivamente em mulheres adultas em idade fértil, contudo homens adultos e crianças também podem ser afetados1-2 e tende a progredir mais rapidamente em jovens e pacientes pré-menopausa.6
Duas formas de LAM estão descritas: a forma esporádica (S-LAM), que ocorre em 3.3-7.7 milhões de mulheres e a forma hereditária autossómica dominante que afeta mulheres com esclerose tuberosa (TSC-LAM).4
Perante o presente caso clínico, o diagnóstico de LAM associada ao complexo de esclerose tuberosa foi assumido pelas alterações quísticas características encontradas na TCAR de tórax e considerando os antecedentes já conhecidos de esclerose tuberosa e a presença de angiomiolipomas renais. Tal como descrito na literatura, o diagnóstico clínico de LAM é estabelecido por quistos redondos bem definidos de paredes finas, cujos tamanhos variam entre milímetros e dois centímetros na TCAR, acompanhado por qualquer das seguintes alterações clínicas: TSC, angiomiolipomas renais, ou derrames pleurais quilosos no tórax ou abdómen.1,7 TSC-LAM é causada pela perda de função de um dos dois genes TSC1 ou TSC2 e caracterizada por crescimentos tumorais tipo em vários órgãos, calcificações cerebrais (como no caso apresentado), convulsões e atraso mental.2,6
Doentes com TSC-LAM são diagnosticadas mais precocemente que as S-LAM e doentes com esclerose tuberosa devem ser rastreadas quanto à existência de LAM para prevenir a rápida deterioração pulmonar.7
A medição sérica de VEGF-D e a constatação da sua elevação superior ou igual 800pg/ml, em especial quando há envolvimento linfático, pode ajudar a estabelecer o diagnóstico de LAM em pacientes sem atingimento extra-pulmonar ou sem TSC e excluir pacientes com outras doenças pulmonares quísticas, evitando assim a necessidade de biópsia pulmonar.1,7 Contudo, valores séricos normais de VEGF-D, como no caso apresentado, não excluem a doença7, mas altos níveis séricos de VEGF-D estão associados a superiores melhorias da função pulmonar quando tratados com sirolimus.1
O pneumotórax ocorre em 50-60% dos pacientes e é mais comum em doentes que apresentem quistos de maior tamanho, podendo ser a manifestação clínica inicial, como no caso apresentado.1,7 Contudo, como se tratava deuma doente fumadora, foi considerada como hipóteseinicial outra doença bolhosa, nomeadamente enfisema secundário à exposição tabágica. Está definido que, logo após o primeiro pneumotórax, seja realizada pleurodesis para prevenir a recorrência.1 No presente caso, a pleurodesis não foi realizada no primeiro episódio, pois só após a resolução do mesmo e a realização de TCAR foi estabelecido o diagnóstico8.
Um padrão funcional ventilatório obstrutivo é mais comum em pacientes em S-LAM (61%) do que em TSC-LAM, mas no caso apresentado a doente evidenciava uma síndrome obstrutiva moderada com resposta positiva ao broncodilatador, o que ocorre em 25 a 30% dos casos, e uma DLCO diminuída, alteração frequente (57%) nestes pacientes.7 Não podemos contudo esquecer o facto de ser fumadora, o que contribui para os achados encontrados nas provas funcionais.
Tal como recomendado, a paciente iniciou terapêutica com sirolimus 1mg/dia.1 O tratamento com Sirolimus mostrou melhorar a função pulmonar e/ou impedir o seu declínio, melhorar o desempenho funcional, a qualidade de vida e reduzir o volume dos angiomiolipomas.1-3 O que verificámos no nosso caso foi uma estabilidade da função pulmonar (avaliada aos 3 e 6 meses após o inicio de terapêutica), uma melhoria da dispneia basal bem como da qualidade de vida, permitindo à paciente voltar à sua atividade laboral. Na maioria das vezes, o sirolimus é bem tolerado e os efeitos adversos são mínimos; contudo, tal como no caso apresentado, podem ocorrer mucosites ou diarreia.1
De referir que a hipercolesterolémia e a hipertrigliceridémia, também presentes na nossa doente, devem ser adequadamente tratadas pois aumentam a probabilidade de desenvolver quilotórax nestes doentes.2
O prognóstico desta patologia é mau e a mortalidade é associada a insuficiência respiratória.2 No momento do diagnóstico nenhum teste é preditivo do prognóstico,7 mas dados clínicos, patológicos e fisiológicos auxiliam na avaliação do prognóstico de pacientes recém-diagnosticadas com LAM.4
O transplante pulmonar é uma das opções terapêuticas para doentes com fim de vida pulmonar e a sobrevida foi considera comparável à dos que realizaram transplante por outras patologias pulmonares terminais.9 Apesar de relatos de que a LAM recorra em pulmões transplantados, consistente com o mecanismo metastático da doença, tal facto não compromete a elegibilidade para o transplante pulmonar até porque as células LAM apresentam características musculares lisas e aspeto histológico benigno.1
A descrição deste caso clínico torna-se pertinente pela raridade desta entidade e gravidade, imagiológica e funcional, representando um desafio no diagnóstico diferencial de doença bolhosa pulmonar em jovem fumadora. Para além disso, o tratamento com sirolimus tornou-se inovador, com estabilidade da função pulmonar, parcos efeitos adversos e melhoria clínica muito significativa na qualidade de vida da paciente.
Figura I
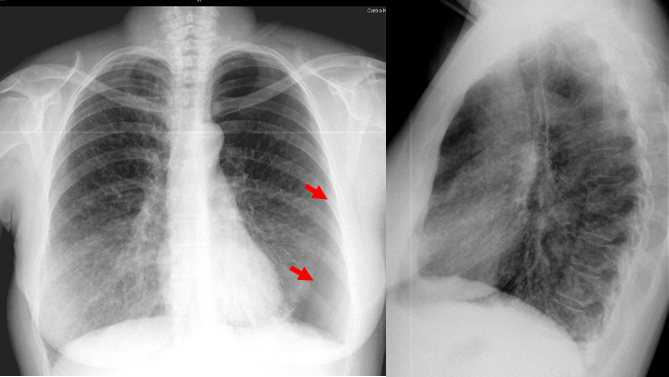
Fig1. Radiografia de tórax à entrada no serviço de urgência presença de padrão intersticial bilateral de predomínio basal e hipertransparência no andar inferior do campo pulmonar esquerdo compatível com pneumotórax espontâneo (setas vermelhas).
Figura II
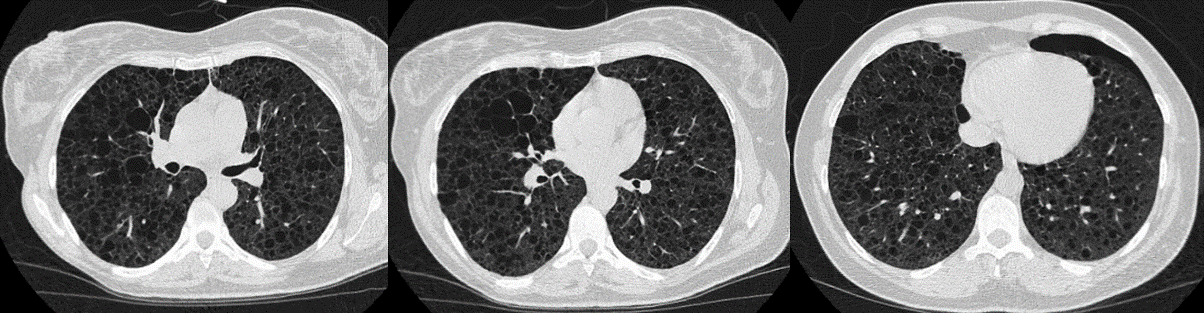
Fig2. TCAR - Inúmeras cavidades quísticas aéreas de parede fina com distribuição bilateral e difusa.
Figura III
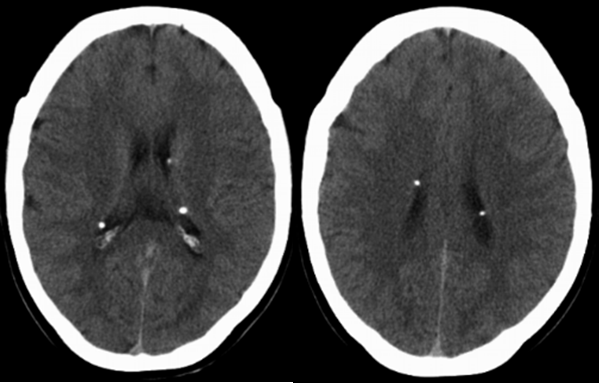
Fig3. TC crânio-encefálica com presença de várias calcificações justa-ventriculares sugestivas de TSC.
BIBLIOGRAFIA
1. McCormack FX, Gupta N, Finlay GR, Young LR, Taveira-DaSilva AM, Glasgow CG, et al. ATS/JRS Committee on Lymphangioleiomyomatosis. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am J Respir Crit Care Med. 2016;194(6):748-61.
2. Tümay LV, Güner OS, Zorluoğlu A. An extrapulmonary manifestation of lymphangioleiomyomatosis: A rare case report. Int J Surg Case Rep. 2017;41:315-8. doi:10.1016/j.ijscr.2017.10.057
3. Zhou B, Guo Q, Zhou H, Xie W, Xue T, Li M, et al. Pulmonary lymphangioleiomyomatosis in a 46-year-old female: A case report and review of the literature. Biomed Rep. 2016 ;4(6):719-22.
4. Taveira-DaSilva AM, Moss J. EPIDEMIOLOGY, PATHOGENESIS and DIAGNOSIS of LYMPHANGIOLEIOMYOMATOSIS. Expert, Opin Orphan Drugs. 2016;4(4):369-78.
5. Pais F, Fayed M, Evans T. Lymphangioleiomyomatosis: an explosive presentation of a rare disease. Oxf Med Case Reports. 2017; 2017(6):omx023.
6. Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035.
7.- Jackson E, Nichani R. Consideration of underlying causes of pneumothoraces in patients presenting with their first pneumothorax. J Intensive Care Soc. 2017;18(4):334-8.
8. Gupta N, Finlay GA, Kotloff RM, Strange C, Wilson KC, Young LR, et al. ATS Assembly on Clinical Problems. Lymphangioleiomyomatosis Diagnosis and Management: High-Resolution Chest Computed Tomography, Transbronchial Lung Biopsy, and Pleural Disease Management. An Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2017; 196(10):1337-1348.
9. Ando K, Okada Y, Akiba M, Kondo T, Kawamura T, Okumura M, et al. Respiratory Failure Research Group of the Japanese Ministry of Health, Labour, and Welfare. Lung Transplantation for Lymphangioleiomyomatosis in Japan. PLoS One. 2016; 11(1):e0146749.

