INTRODUÇÃO
A Linfohistiocitose hemofagocítica (LHH) é uma entidade rara, que envolve uma resposta descontrolada do sistema imune com produção exagerada de citoquinas pro-inflamatórias, levando a disfunção multiorgânica, que se traduz numa elevada taxa de mortalidade (1, 2). Pode ser de etiologia primária, tendo por base alterações genéticas, ou secundária, como por exemplo, por infeção, neoplasias ou doenças autoimunes (1,2). A clínica inespecífica e comum a muitas outras patologias torna o diagnóstico difícil e desafiante. Apesar do nome dado a esta síndrome, de salientar que a hemofagacitose pode não estar presente em até 30% dos casos, e estando presente, por si só não faz o diagnóstico, uma vez que a hemofagocitose medular pode ocorrer em doentes sem LHH, devendo-se a outras entidades (2).
CASO CLÍNICO
Os autores descrevem o caso clínico de uma mulher de 84 anos, autónoma, que desenvolveu um quadro de astenia e prostração progressivas, com perda de autonomia, com evolução de 2 semanas. Tinha hipertensão arterial essencial sem lesões de órgão alvo conhecidas, glaucoma e patologia osteoarticular degenerativa. Sem antecedentes familiares de relevo.
Na admissão no serviço de urgência apresentava-se desorientada, sem sinais meníngeos, febril, hipotensa e com taquicardia sinusal. Gasimetria com insuficiência respiratória tipo 1 (pO2/FiO2 260) e hiperlactacidemia (5mmol/L). Analiticamente apresentava bicitopenia grave - anemia aguda inflamatória (5,4g/dL), coombs positiva por complemento, com hemólise extravascular (desidrogénase láctica de 600) e trombocitopenia (29000/uL). Destacava-se ainda elevação da proteína C reativa (329mg/L) e da velocidade de sedimentação (99mm). O estudo imagiológico documentou hepatoesplemegalia maciças (20cm), sem outras alterações de relevo, nomeadamente adenopatias.
Excluída infeção respiratória, abdominal e do sistema nervoso central. Documentada bacteriúria a Escherichia coli que foi tratada com antibioterapia dirigida, sem melhoria do quadro clínico. Etiologia vírica descartada, nomeadamente serologias de HIV, EBV, CMV e hepatites B, C e E negativas.
O estudo analítico complementar evidenciou hiperferritinemia ˃ 6000 ng/mL, hipertrigliceridemia ˃ 400 mg/dL, hipofibroginemia e défice de IgG. Assumida LHH, cumprindo os critérios de HScore - Fardet et al (3) -, contabilizando 263 pontos. Iniciou reposição de imunoglobulinas e prednisolona 1mg/kg, mantendo evolução clínica desfavorável com persistência de febre e resposta inflamatória sistémica exuberante.
Prosseguiu-se investigação com estudo medular que documentou imagens de hemofagocitose (principalmente de leucócitos) e envolvimento medular por doença linfoproliferativa B com características fenotípicas sugestivas de linfoma B de grandes células, com expressão de CD30 (Fig 1 A-E).
Persistência de deterioração clínica rapidamente progressiva e atendendo à idade avançada, com prognóstico muito reservado, foi decidido em equipa multidisciplinar, não ter benefício em iniciar tratamento dirigido ao linfoma, nomeadamente quimioterapia. A doente veio a falecer ao 10º dia de internamento.
DISCUSSÃO
A LHH é uma síndrome potencialmente ameaçadora de vida, que pode ter uma evolução indolente ou progressiva e catastrófica. Caracteriza-se por desregulação do sistema imunológico, com produção excessiva de citoquinas, provocando um estado inflamatório extremo e lesão dos tecidos com falência multiorgânica e elevada mortalidade que pode atingir os 40% (1, 2, 4). Há critérios definidos para diagnóstico desta síndrome, mas que se sobrepõem a outras entidades, o que o torna difícil o seu reconhecimento, obrigando a um elevado índice de suspeição, e consequentemente a um diagnóstico tardio, com início de tratamento em fases avançadas da doença, com impacto no prognóstico.
A LHH pode ser primária ou secundária a infeção, neoplasias ou doenças autoimunes, sendo as secundárias as mais comuns (1). Entre as causas neoplásicas, pode associar-se à própria neoplasia ou aos tratamentos da mesma, e as mais frequentemente envolvidas, são as neoplasias hematológicas, nomeadamente linfomas (1, 5, 6, 7), como o caso descrito pelos autores.
A expressão clínica da tempestade pro-inflamatória que ocorre na LHH, caracteriza-se por febre, citopenias e disfunção de órgão, a maioria das vezes multiorgão (1, 2). Os critérios de diagnóstico mais recentes HScore (Fardet et al. 3) englobam a febre, hepatoesplenomegalia, citopenias, hipertrigliceridemia, hipofibrinogemia, hemafagocitose no aspirado medular, hiperferritinemia, elevação das transamínases ou a existência de imunossupressão prévia. HScore > 169 apresenta uma sensibilidade de 93% e especificidade de 86%, no diagnóstico desta síndrome (1).
Não há estudos randomizados sobre o tratamento, sendo que a maioria dos protocolos recaem sobre a terapêutica imunossupressora utilizada na LHH primária (1). Por isso, torna-se imperativo que o diagnóstico seja o mais precocemente possível, com o suporte de órgão adequado e identificação e tratamento da causa subjacente, enquanto se inicia a terapêutica imunossupressora para controlo da desregulação inflamatória (1). O tratamento deve ser discutido em equipa multidisciplinar, por ser uma doença rara e de diagnóstico difícil. O reconhecimento do trigger das formas secundárias é fundamental, uma vez que o tratamento da LHH pode interferir com a doença causal.
Os corticoides são considerados a terapêutica imunossupressora primária, mas podem mascarar algumas entidades, como por exemplo, os linfomas (1). O Anakinra é um antagonista do recetor da IL-1 recombinante, efetivo no controlo da tempestade pro-inflamatória, sendo bem-tolerado e com perfil de efeitos-laterais favorável que tem ganho destaque, principalmente na LHH secundária a patologia autoimune ou idiopática (1).
O caso descrito, reflete uma doente com sintomatologia inespecífica, mas objetivamente com indicadores de gravidade, apresentando disfunção multiorgânica. Outros diagnósticos podiam ser considerados, entre os quais a sépsis. Contudo, a gravidade das citopenias e a ausência de melhoria clínica da doente sob antibioterapia eficaz, levaram à célere perseguição deste diagnóstico. Os autores apresentaram um caso clínico com um HScore elevado (263 pontos) a favorecer o índice de certeza do diagnóstico, e uma das causas mais frequentemente envolvidas na LHH secundária o linfoma. Contudo, salienta-se a raridade deste diagnóstico, principalmente atendendo à idade de apresentação. A terapêutica com corticoide foi iniciada empiricamente, ainda antes do diagnóstico definitivo, em dose inferior à recomendada para não mascarar o diagnóstico definitivo. Outras terapêuticas dirigidas ao linfoma ou à LHH, nomeadamente quimioterapia, não foram consideradas, atendendo ao estado clínico da doente, com deterioração progressiva e atingimento multiorgânico, sem resposta à terapêutica imunossupressora inicial. Considerou-se que o risco/benefício de iniciar tratamento dirigido à doença de base não beneficiava em nada o bem-estar nem o prognóstico da doente, atendendo à baixa reserva fisiológica que apresentava.
Este caso demonstra, a progressão rápida e catastrófica que frequentemente se associa a esta síndrome, agravando significativamente o prognóstico dos doentes.
Os autores pretendem alertar para o diagnóstico desta entidade rara, que se pode apresentar em qualquer idade, e que se não for rapidamente equacionada, facilmente se perde entre sintomas inespecíficos que percorrem muitas outras entidades clínicas, sem permitir um tratamento atempado e eficaz.
Figura I
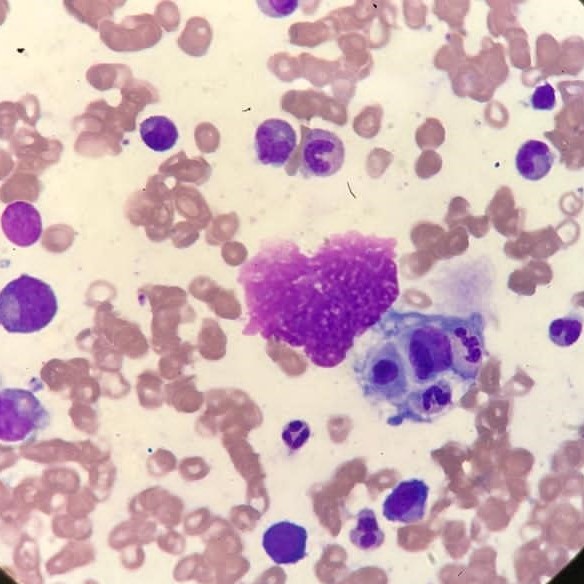
Fagocitose de pequenos linfócitos e granulócitos - Imagem de microscopia ótica de amostra de medula óssea (coloração de Leishman)
Figura II
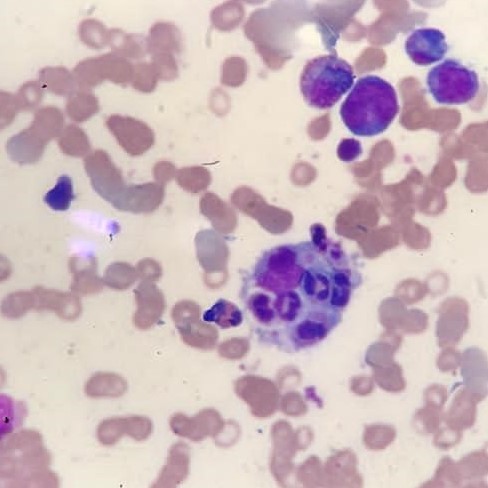
Fagocitose de neutrófilos - Imagem de microscopia ótica de amostra de medula óssea (coloração de Leishman)
Figura III
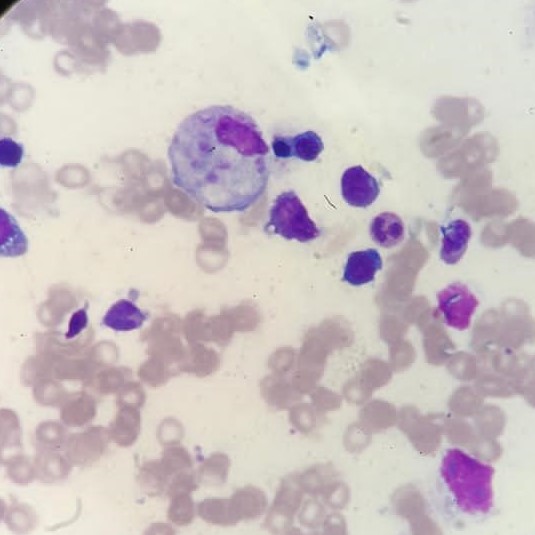
Fagocitose de plaquetas - Imagem de microscopia ótica de amostra de medula óssea (coloração de Leishman)
Figura IV
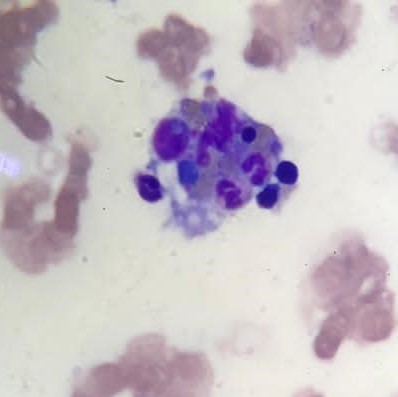
Granulócitos, pequenos linfócito e eritroblastos a serem internalizados por fagócito - Imagem de microscopia ótica de amostra de medula óssea (coloração de Leishman)
Figura V
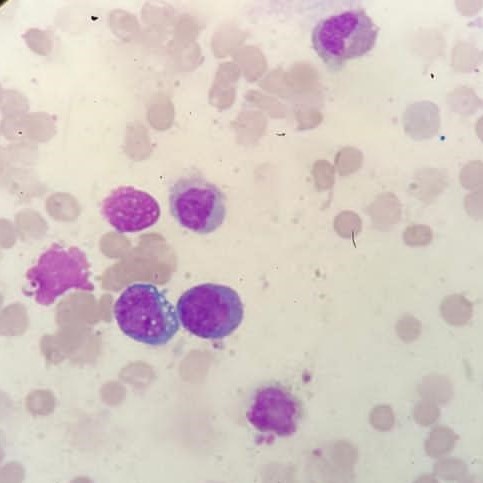
Linfócitos atípicos com citoplasma basófilo com alteração da razão núcleo citoplasma e presença de múltiplos vacúolos - Imagem de microscopia ótica de amostra de medula óssea (coloração de Leishman)
BIBLIOGRAFIA
1. Bauchmuller K, Manson J J, Tattersall R, Brown M, McNamara C, Singer M, et al. Haemophagocytic lymphohistiocytosis in adult critical care. Journal of the Intensive Care Society. 2020; 21(3): 25668
2. Hutchinson M, Tattersall R, Manson J J. Haemophagocytic lymphohisticytosisan underrecognized hyperinflammatory syndrome. Rheumatology. 2019; 58: vi23vi30
3. Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014; 66: 261320
4. Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta M, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014; 383: 150316
5. Daver N, McClain K, Allen CE, Parikh S, OtrockZ, Rojas-Hernandez C, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017; 123: 322940
6. Delavigne K, Berard E, Bertoli S, Corre J, Duchayne E, Demur C, et al. Hemophagocytic syndrome in patients with acute myeloid leukemia undergoing intensive chemotherapy. Haematologica. 2014; 99: 47480
7. Machaczka M, Vaktnas J, Klimkowska M, Hagllund D. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: A retrospective population-based analysis from a single center. Leukemia Lymphoma. 2011; 52: 6139.

