INTRODUÇÃO
A Doença de Anderson-Fabry, também conhecida como Doença de Fabry (DF) ou Angiokeratoma Corporis Diffusum Universale, é uma doença rara de sobrecarga lisossomal.1 Esta resulta da mutação no gene da enzima alfa-galactosidase A (GLA), localizado no cromossoma X (Xq22.1), condicionando diminuição ou ausência da enzima e consequente acumulação de glicoesfingolípidos nos lissosomas, como a globotriaosilceramida (GL-3 ou Gb3), particularmente nas células endoteliais, neuronais, cardíacas e renais, com lesão progressiva e irreversível de órgão.2-4
A doença é pan-étnica e tem uma prevalência estimada de um em cada 40 000 a 60 000 indivíduos do sexo masculino, sendo este número provavelmente subestimado devido à existência de formas assintomáticas, moderadas e tardias da doença.5
A complexidade da doença resulta de diversos fatores, como o facto de existirem mais de 1000 mutações descritas do gene GLA, da heterogeneidade fenotípica dependente da atividade residual da enzima e da ausência de correlação genótipo-fenotípica que determine risco clínico.1,3
A apresentação clássica da doença ocorre geralmente em homens hemizigotos. As mulheres heterozigotas, embora possam ser tão gravemente afetadas como os homens, tendem a ter uma apresentação mais tardia e insidiosa.3
CASO CLÍNICO
Descrevemos o caso de uma mulher de 47 anos, leucodérmica, com história pessoal de amaurose do olho direito devida a descolamento da retina por catarata congénita; hipotiroidismo; ansiedade e tabagismo (20UMA). Como antecedentes familiares destaca-se como relevante o falecimento do tio materno aos 50 anos por acidente vascular cerebral (AVC), negando história familiar de outras patologias do foro cardiovascular e renal. A doente tem 2 filhos, uma filha com 18 anos de idade e um filho com 22 anos, ambos aparentemente saudáveis.
Seguida em consulta de risco cerebrovascular desde os 35 anos após AVC isquémico, com hemiparesia direita e ligeira disartria. Realizou tomografia computorizada crânio-encefálica (TC-CE) que documentou enfarte lenticulo-capsular posterior esquerdo não recente e ressonância magnética crânio-encefálica (RM-CE) que confirmou a lesão. Pedido estudo etiológico exaustivo para AVC no jovem que foi inconclusivo. Iniciou prevenção secundária com estatina e antiagregante plaquetarário, mantendo seguimento regular em consulta. Desde os 36 até aos 41 anos apresentou episódios recorrentes e autolimitados de hipoestesia da mão e hemiface, que alternavam bilateralmente. Nesta fase realizou eletroencefalograma e electromiografia que não demonstraram alterações e repetida TC-CE sem documentação de lesão de novo. Aos 42 anos, recorre novamente ao serviço de urgência por dormência da hemiface e membro superior esquerdos, com diminuição da força muscular homolateral com um dia de evolução. Ao exame objetivo apresentava apagamento do sulco nasogeniano direito e hemiparesia direita de grau 3+, ambos sequelares, e de novo, hemihipostesia álgica da face e membro superior à esquerda, hemiparésia esquerda de predomínio braquiofacial força muscular grau 4+, e marcha atáxica. Realizado estudo imagiológico do qual se destaca RM-CE com múltiplas lesões isquémicas bilaterais, sem evidência de dolicoectasia arterial. (Fig. 1). Observada novamente em consulta, já com importante limitação da mobilidade, com marcha possível apenas com auxílio de canadianas. Repetido estudo exaustivo para AVC no jovem destacando-se como única alteração uma microalbuminúria de 287,9 mg/dL, com restante estudo uma vez mais inconclusivo. Implantou-se registador de eventos, que não apresentou registos entre os 42 e os 45 anos. Dada a gravidade de apresentação de doença cerebrovascular com envolvimento multifocal, ponderada a hipótese de doença genética. Estudo CADASIL com sequenciação dos exões 2-6 e 11 do gene NOTCH3, sem mutações. Estudo para doença de Fabry com genotipagem do gene GLA que detetou a presença, em heterozigotia, da variante genética c.1229C>T, descrita como causal da Doença de Fabry e doseamento da atividade enzimática da alfa-galactosidase A com valor normal no plasma 15 nmol/h/ml (valor de referência 6-19) e valor abaixo do intervalo de referência nos leucócitos 29 nmol/h/mg (valor de referência 36-80). Doente sem outros sinais ou sintomas, nomeadamente, angioqueratomas, acroparestesias, córnea verticilata ou crises álgicas. Referenciada para nefrologia, sendo realizada biópsia renal que demonstrou, por microscopia ótica, arquitetura relativamente conservada com alterações discretas que podem ser enquadradas no contexto de Doença de Fabry - presença de 5 de 21 glomérulos escleróticos, fibrose intersticial e atrofia tubular e arterioesclerose ligeiras. Nesta fase doseamento de Gb3 e globotriaosilesfingosina (Lyso Gb3) plasmáticos e Gb3 urinário negativos. RM cardíaca demonstrou: VE não dilatado, com hipertrofia do septo anterior basal (12 mm) sem alteração da mobilidade segmentar, com boa função sistólica, sem déficits de perfusão e sem focos de realce tardio. Dado fenótipo de predomínio cerebrovascular grave com morbilidade muito importante, doente foi proposta para terapêutica de reposição enzimática, que foi recusada, tendo em conta a irreversibilidade das lesões cerebrovasculares, o facto desta terapêutica não ultrapassar a barreira hematoencefálica e ausência de compromisso de outros órgãos e sistemas à data da proposta. Sendo a mutação da doente (variante genética c.1229C>T) descrita como suscetível para migalastate, foi proposta posteriormente e aceite para esta terapêutica, que iniciou há 1 ano.
Aos 46 anos teve dois internamentos consecutivos no Serviço de Neurologia por neuropatia ótica isquémica anterior (NOIA) no contexto de doença vascular secundária à DF, com perda irreversível da acuidade visual. No decorrer dos seus 46 anos de idade, a doente apresentou envolvimento de outros órgãos, nomeadamente cardiovascular, com necessidade de implantação de pacemaker por episódios recorrentes de pausas prolongadas, e renal, com agravamento progressivo de proteinúria (600mg/24h). Nesta fase doseamento de Gb3 e Lyso Gb3 plasmáticos e Gb3 urinário elevados.
DISCUSSÃO
A DF é uma doença de armazenamento lisossomal, que poderá acometer vários órgãos e sistemas, nomeadamente, cutâneo, cardiovascular, renal, gastrointestinal e cerebrovascular. Na sua forma clássica, a doença pode manifestar-se desde a infância/adolescência com angioqueratomas, acroparestesias, sintomas gastrointestinais (dor abdominal pós-prandial e distensão abdominal, saciedade precoce, diarreia, náuseas e vómitos), intolerância ao calor, frio e exercício físico, hipotensão ortostática e hipo ou hiperhidrose.2-4 Podem ocorrer ainda crises álgicas de Fabry induzidas pelo stress, exercício físico, calor e frio.2-4 Outra manifestação muito frequente é a córnea verticilata, presente em cerca de 90% dos doentes, e que raramente afeta a visão. 2-4 Na idade adulta predominam as manifestações cardíacas, cerebrovasculares e renais.3,4
Em relação às manifestações renais, a proteinúria e declínio da taxa de filtração glomerular surgem por volta da 2ª a 3ª década de vida, o que nos homens pode evoluir para DRC terminal por volta da 4ª a 5ª década de vida.2,3
As manifestações cardiovasculares estão presentes em cerca de 40 a 60% dos doentes, podendo ocorrer hipertrofia ventricular esquerda, insuficiência valvular aórtica e mitral, alterações da condução, doença coronária microvascular, dilatação da raíz da aorta e hipertrofia do ventrículo direito.2,3
As complicações cerebrovasculares incluem acidentes isquémicos transitórios, AVCs isquémicos e hemorrágicos e lesões da substância branca. O AVC afeta 6.9% dos homens com idade média de 39 anos, podendo ser a primeira manifestação de DF em 4.3% dos doentes. 2,3
Nos fenótipos atenuados, a apresentação é mais tardia, e envolve geralmente apenas um órgão (fenótipos cardíaco, renal ou cerebrovascular).3,4
Nas mulheres heterozigotas o espectro de apresentação é amplo, e geralmente apresentam a doença de forma mais tardia e insidiosa, o que poderá estar relacionado com a inativação aleatória do cromossoma X.3,4
No caso da doente que apresentamos, a primeira manifestação da doença foi um AVC isquémico, cujo estudo complementar etiológico se demonstrou inconclusivo, não se colocando à data da apresentação a possibilidade de causa genética, dada ausência de outra sintomatologia associada que orientasse o diagnóstico. A doente descrita apresenta um fenótipo cerebrovascular severo, com uma morbilidade muito acentuada, sendo o diagnóstico realizado cerca de 10 anos após o primeiro evento.
O diagnóstico da DF pode efetuar-se pela sequenciação do gene GLA e/ou pela determinação da atividade da α-galactosidase A. No caso dos hemizigotos, a determinação da atividade da α-galactosidase A (através de testes de gota seca [dried blood spot DBS], no plasma, leucócitos, fibroblastos ou em tecidos biopsados) constitui a primeira escolha na avaliação dos doentes suspeitos.3,7 Nos homens com fenótipo fenótipo atenuado, pode ser necessária a sequenciação do gene GLA para diagnóstico definitivo.3,7 No diagnóstico dos heterozigotos a sequenciação do gene GLA é obrigatória, dado que estas podem apresentar níveis de atividade enzimática dentro da normalidade.3,7
Desde 2001 para além da terapêutica sintomática, passou a estar disponível terapêutica de reposição enzimática (TRE) - agalsidase alfa e agalsidase beta - que demonstrou aliviar significativamente os efeitos cardíacos, renais e neuropáticos, com impacto na progressão da doença e na qualidade de vida.6 Desde 2018, encontra-se disponível em Portugal, a primeira chaperona oral o migalastate que atua ligando-se e estabilizando determinadas formas mutantes da enzima α-Gal A, cujos genótipos são descritos como mutações do GLA suscetíveis, de forma a potenciar a sua atividade.8 A nossa doente encontra-se medicada desde há 1 ano com migalastate, após ter sido proposta e recusada para TRE.
O diagnóstico da doente, permitiu ainda iniciar o rastreio familiar. O filho na altura com 19 anos, e até à data assintomático, é portador da mutação variante c.1229C>T do gene GLA, com doseamento da alfa-galactosidase A por DBS de 0,39pmol/h/punção (valor de referência 8,75-15,6). À filha,na altura menor de idade e não residente no país, foi protelado rastreio até à maioridade por decisão parental.
A DF quando não tratada associa-se a uma redução de sobrevida em cerca de 20 anos nos homens e 10 anos nas mulheres2, pelo que o diagnóstico atempado permitirá não só o início de terapêutica precoce, como o rastreio familiar e devido aconselhamento genético que poderão condicionar o curso natural da doença e a sua morbimortalidade.
AGRADECIMENTOS
Os autores agradecem a colaboração das colegas Dra. Pureza Mateus, Serviço de Neurologia do Centro Hospitalar de Leiria, e da Dra. Maria Guedes, Serviço de Nefrologia do Centro Hospitalar Universitário de Coimbra Hospital dos Covões, por toda a disponibilidade e colaboração. Agradecem ainda à Dra. Maria de Jesus Banza, Diretora do Serviço de Medicina do Centro Hospitalar de Leiria.
Consentimento para Publicação
Foi obtido consentimento informado da doente para publicação deste caso clínico.
Figura I
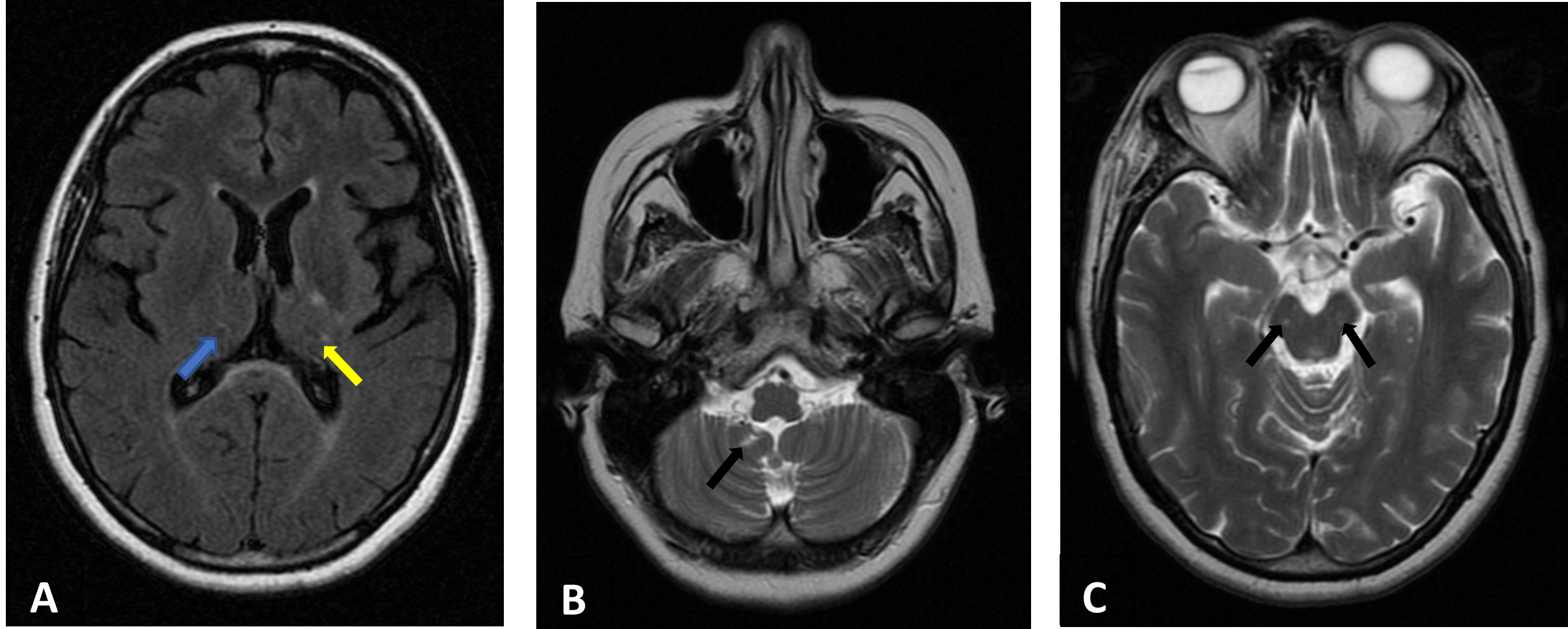
RM-CE: Enfarte isquémico sequelar lenticulo-capsular posterior e talâmico esquerdo (A seta amarela); enfarte sub-agudo lenticulo-capsular posterior e talâmico direito (A seta azul), enfarte sequelar antero-inferior cerebeloso direito (B seta preta); lacunas isquémicas protuberanciais bilaterais (C setas pretas) - Sequências axiais T2 (A) e FLAIR (B) e (C).
BIBLIOGRAFIA
REFERÊNCIAS:
1- Boggio P, Luna PC, Abad ME, Larralde M. Doença de Fabry [Fabry disease]. An Bras Dermatol. 2009 Jul-Aug;84(4):367-76
2- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30.
3- Beirao I, Cabrita A, Torres M, Silva F, Aguiar P, Gomes AM. Será Doença de Fabry? Abordagem Diagnóstica e de Seguimento [Is it Fabry Disease? Diagnostic and Follow-Up Approach]. Acta Med Port. 2016 Feb;29(2):85
4- Eng CM, Germain DP, Banikazemia M, Warnock D, Wanner C, Hopkin RJ, et al. Anderson-Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Gent Med. 2006;9:539-48.
5- DESNICK RJ, BRADY R, BARRANGER J et al: Fabry Disease, an Under-Recognised Multisystemic Disorder: Expert Recommendations, for Diagnosis, Management, and Enzyme Replacement, Therapy. Ann Intern Med 2003;138:338-346
6- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416427
7- Gal A, Hughes DA, Winchester B. Toward a consensus in the laboratory diagnostics of Fabry Disease recommendations of an European expert group. J Inherit Metab Dis. 2011;34:509-14.
8- McCafferty EH, Scott LJ. Migalastat: A Review in Fabry Disease. Drugs. 2019 Apr;79(5):543-554

