

Introdução
A Macroglobulinemia de Waldenstrom (MW) é um distúrbio linfoproliferativo de células B. Define-se por uma infiltração clonal linfoplasmocítica da medula óssea ou do tecido linfóide associada a uma proteína IgM monoclonal no soro.1,2 É uma doença rara e os sintomas variam consideravelmente.
Caso Clínico
Um homem de 84 anos, caucasiano, autónomo, recorreu ao Médico de Família por dispneia para médios esforços, tosse não produtiva, astenia e anorexia com um mês de evolução. Na radiografia torácica, apresentava uma hipotransparência na base pulmonar direita. Foi medicado com levofloxacina durante 10 dias. Por ausência de melhoria, realizou tomografia computorizada (TC) torácica que mostrava derrame pleural à direita com áreas de consolidação, um conglomerado adenopático com 4 cm ao longo do brônquio principal direito e estruturas ganglionares com aspeto irregular. Perante este resultado, foi enviado ao Serviço de Urgência de Medicina Interna.
Tinha antecedentes de hipertensão arterial, gastropatia erosiva, meningioma e hipertrofia benigna da próstata. Trabalhava como pedreiro, com exposição ocupacional ao pó da pedra. Sem história familiar relevante.
Ao exame objetivo à admissão, encontrava-se hemodinamicamente estável e apirético. Na auscultação pulmonar, apresentava diminuição do murmúrio vesicular nas bases, mais evidente à direita, e crepitações bibasais. Sem outras alterações de relevo, nomeadamente adenomegalias ou organomegalias. Do estudo realizado salientava-se: hemograma sem alterações, proteína C reativa negativa, função renal, ionograma, provas de função e lesão hepática normais. Repetiu TC torácica, onde se observava derrame pleural direito de moderado volume, com atelectasia parcial do lobo inferior homolateral, sem áreas de consolidação ou outras lesões (Fig. 1). Estava presente uma adenomegalia infracarinal de 17 mm. Identificavam-se várias formações ganglionares mediastínicas, infra e pericentimétricas, sem características de adenopatia, não sendo visível o conglomerado adenopático descrito no relatório de TC tórax realizado no exterior. Foi realizada toracocentese diagnóstica. O líquido pleural foi compatível com um exsudado com predomínio de mononucleares, com adenosina desaminase de 61,1 U/L (N< 60).
Ficou internado no Serviço de Medicina Interna para investigação diagnóstica. Foram colocadas como principais hipóteses de diagnóstico tuberculose e doença neoplásica (tumor primário do pulmão ou da pleura, doença metastática ou distúrbio linfoproliferativo).
Durante o internamento, não apresentou outras queixas. Manteve-se hemodinamicamente estável, apirético, sem necessidade de oxigenoterapia e com exame objetivo sobreponível ao descrito. A análise microbiológica direto do líquido pleural foi negativa. A citologia revelou células mesoteliais reativas e numerosas células inflamatórias de predomínio mononucleado. Programou-se biópsia pleural e colheita de líquido para imunofenotipagem. Contudo, a radiografia torácica prévia ao procedimento revelou o desaparecimento total do derrame, pelo que não foi possível a sua realização. Posteriormente, a pesquisa por PCR de DNA de Mycobacterium tuberculosis no líquido pleural foi positiva. Foi iniciado tratamento para tuberculose pleural com isoniazida, rifampicina, etambutol e pirazinamida. A serologia do vírus da imunodeficiência humana era negativa. O doente cumpriu 2 meses de terapêutica antibacilar quádrupla seguido de 4 meses de isoniazida e rifampicina, com resposta favorável. Não foi houve reaparecimento de derrame pleural nem evidência de adenomegalias mediastínicas em TC torácicas subsequentes.
Durante o internamento, foi ainda documentado um pico de base estreita na região gama da eletroforese de proteínas séricas. O estudo foi, então, dirigido para a pesquisa de discrasia plasmocitária. Verificou-se elevação da IgM (1790 mg/dL), diminuição da IgA (40,5 mg/dL) e da IgG (559 mg/dL). A imunoeletroforese de proteínas séricas mostrava uma banda de precipitação monoclonal do tipo IgM cadeias leves kappa, com relação kappa/lambda de 4,21. Não foram detetadas cadeias leves por electroimunofixação na urina de 24 horas. A β2-microglobulina era de 3,2 mg/L (N<0,253). As radiografias de ossos longos e calote craniana não revelaram lesões líticas. O mielograma apresentava 0,7% de plasmócitos. A imunofenotipagem de sangue medular identificou uma população de linfócitos B monoclonal com expressão de cadeias leves kappa, com características compatíveis com linfoma linfoplasmocítico (CD19+/CD20+/CD5-/CD10-/CD23-/FMC7+/CD79b+/CD11cvariável/CD25+/CD103-/CD38variável/IgM+/cadeias leves kappa+). A população monoclonal representava 96% das células B, 36% das células linfoides e 6,1% de todas as células do aspirado medular. Foi assim firmado o diagnóstico de MW. Para avaliação do prognóstico foi utilizado o International Prognostic Scoring System for Waldenstrom Macroglobulinemia (MW IPSS), no qual o doente pontuava 2. Correspondia a um risco intermédio com sobrevida média estimada em 98,6 meses. Atendendo à ausência de complicações relacionadas com infiltração da medula óssea ou com os níveis circulantes de IgM, foi decidido não iniciar tratamento dirigido e manter vigilância. Após 3 anos, o doente encontrava-se assintomático, sem qualquer manifestação da doença hemato-oncológica, com pico M estável (2000 mg/dL na última consulta).
Discussão
A MW é uma doença rara, que representa 1 a 2% de todos os linfomas não Hodgkin.1 É mais frequente na raça caucasiana, sexo masculino e idosos.1-4 Na Europa, a incidência ajustada à idade é de 7,3/1.000.000 no sexo masculino e 4,2/1.000.000 no sexo feminino.2,5 A idade média ao diagnóstico varia entre 63 e 75 anos.1-3,6 A ocorrência familiar é comum mas as bases moleculares permanecem desconhecidas.2,4 Os familiares de 1º grau dos doentes afetados têm risco até 20 vezes superior de desenvolver MW.7
Cerca de 25% dos doentes estão assintomáticos ao diagnóstico.1 Quando presentes, os sintomas podem estar relacionados com os elevados níveis circulantes de IgM ou com infiltração da medula óssea ou outros tecidos por células B malignas. A manifestação clínica major é a síndrome de hiperviscosidade, resultante da presença da proteína IgM monoclonal. Pode manifestar-se através de hemorragia das mucosas, sintomas neurológicos, sintomas constitucionais e sintomas cardiorrespiratórios.1-4 A infiltração dos tecidos pelas células malignas pode resultar em hepatomegalia, esplenomegalia e adenomegalias.2 A anemia é comum e pode ter causa multifatorial: diminuição da produção eritrocitária por infiltração da medula óssea, podendo ser acompanhada de neutropenia e trombocitopenia; inflamação crónica com aumento da produção de hepcidina; aumento da destruição eritrocitária no contexto de anemia imunohemolítica.1,3 A polineuropatia inflamatória desmielinizante crónica ocorre quando a proteína IgM monoclonal apresenta especificidade para a glicoproteína associada a mielina, desencadeando um processo de desmielinização, que pode preceder o aparecimento da neoplasia1,3,7 Outras complicações mais raras associadas à presença de proteína IgM monoclonal são a amiloidose e a crioglobulinemia mista tipo 2.3 Ao contrário do mieloma múltiplo, a MW não está associada a lesões líticas nem hipercalcemia e a excreção renal de proteína monoclonal é baixa; por estes dois motivos, a doença renal não é comum.4
O diagnóstico de MW é estabelecido na presença de uma proteína monoclonal IgM no soro associada a infiltração linfoplasmocítica da medula óssea.2 Em 80% dos casos, o isotipo de cadeia leve é kappa.4 A pesquisa da mutação MYD88 L265P nas células linfoplasmocíticas pode ser útil para diagnóstico diferencial com outras doenças morfologicamente semelhantes como o mieloma múltiplo, estando presente em 90% dos doentes com MW.2-4
Os doentes assintomáticos não têm indicação para iniciar tratamento, devendo manter vigilância. O tempo médio desde o diagnóstico até ao aparecimento de sintomas pode exceder os 5-10 anos.2 As indicações para tratamento incluem a presença de sintomas B, citopenias, hiperviscosidade, neuropatia, amiloidose, crioglobulinemia sintomática ou doença das aglutininas frias.2 A primeira linha de tratamento consiste em combinações de rituximab com agentes alquilantes, inibidores do proteassoma ou inibidores da tirosina cinase de Bruton. Estes últimos também podem ser usados em monoterapia.9 A plasmaferese deve ser usada para alívio imediato dos sintomas de hiperviscosidade, associada a terapêutica sistémica.2,9
O IPSS é um score validado para estratificação de risco na MW sintomática, que considera como fatores de mau prognóstico idade ≥ 65 anos, hemoglobina ≤ 11,5 g/dL, plaquetas ≤ 100x109/L, β2 microglobulina > 3 mg/L e proteína monoclonal sérica > 70 g/L.2
A MW é, geralmente, indolente, com uma sobrevida média que pode exceder os 10 anos em doentes com menos de 70 anos e de cerca de 4 anos em doentes com mais de 80 anos.2-3 Estudos prévios revelaram que a causa de morte em doentes com mais de 75 anos só em 40% dos casos é atribuível à MW.10 Portanto, nos doentes mais velhos, a decisão de instituir terapêutica potencialmente tóxica deve ter em conta o seu efeito na sobrevida e qualidade de vida do doente.
Apresentamos o caso de um homem caucasiano de 84 anos com uma MW de cadeias leves kappa, diagnosticada acidentalmente no decurso do estudo de um derrame pleural, que se verificou ser de etiologia bacilar. Estudos prévios demonstraram que alguns tipos de infeção estão associados a risco aumentado de desenvolver MW, mas não há uma associação estabelecida com tuberculose.11-12 Por outro lado, está descrito que doentes com MW têm risco 3,4 vezes superior de desenvolver infeção de qualquer tipo. Este risco é maior no primeiro ano após o diagnóstico, nos homens e aumenta com a idade.13 São raros os casos descritos de tuberculose em doentes com MW, não havendo dados sobre o risco de tuberculose nestes doentes, sobretudo nos que não estão sob quimioterapia, que provoca imunossupressão por si só. Durante o tratamento da tuberculose, o doente ficou assintomático e houve regressão da adenopatia evidente na TC torácica, pelo que se atribuem os sintomas respiratórios e constitucionais e a adenopatia à tuberculose e não à MW.
Trata-se, então, de um homem com MW indolente, sem indicação para tratamento, que mantém seguimento há 3 anos em Consulta de Medicina Interna.
Figura I
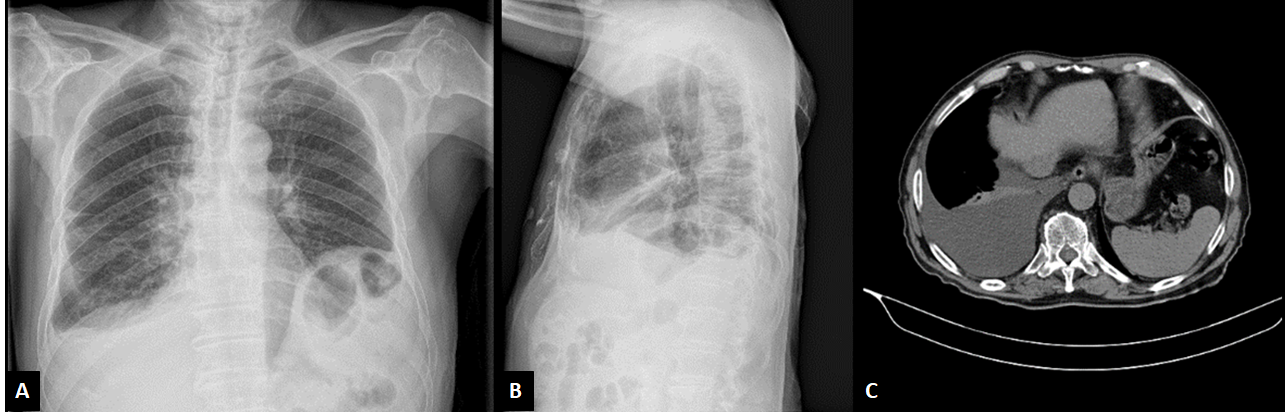
Radiografia de tórax em postero-anterior (A) e perfil (B) e tomografia axial computorizada torácica (C) na admissão do doente no Serviço de Urgência.
BIBLIOGRAFIA
1. Kapoor P, Ansell SM, Fonseca R, Chanan-Khan A, Kyle RA, Kumar SK, et al. Diagnosis and Management of Waldenström Macroglobulinemia: Mayo Stratification of Macroglobulinemia and Risk-Adapted Therapy (mSMART) Guidelines 2016. JAMA Oncol. 2017;3(9):1257-1265. doi:10.1001/jamaoncol.2016.5763.
2. Kastritis E, Leblond V, Dimopoulos MA, Kimby E, Staber P, Kersten MJ, et al. Waldenström´s macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl4):iv41-iv50. doi:10.1093/annonc/mdy146.
3. Gertz MA. Waldenström macroglobulinemia: 2019 update on diagnosis, risk stratification, and management. Am J Hematol. 2019;94(2):266-276. doi:10.1002/ajh.25292.
4. Munshi NC, Longo DL, Anderson KC. Plasma Cell Disorders. In: Jameson JL, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J. Harrison´s Principles of Internal Medicine. New York: McGraw Hill Education; 2018. p.793-803.
5. Phekoo KJ, Jack RH, Davies E, Møller H, Schey SA. The incidence and survival of Waldenström´s Macroglobulinaemia in South East England. Leuk Res. 2008;32(1):55-9. doi:10.1016/j.leukres.2007.02.002.
6. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459. Doi:10.3322/caac.21357.
7. Kristinsson SY, Björkholm M, Goldin LR, McMaster ML, Turesson I, Landgren O. Risk of lymphoproliferative disorders among first-degree relatives of lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia patients: a population-based study in Sweden. Blood. 2008;112(8):3052-6. doi:10.1182/blood-2008-06-162768.
8. Chaudhry HM, Mauermann ML, Rajkumar SV. Monoclonal Gammopathy-Associated Peripheral Neuropathy: Diagnosis and Management. Mayo Clin Proc. 2017;92(5):838-850. doi:10.1016/j.mayocp.2017.02.003.
9. Castillo JJ, Advani RH, Branagan AR, Buske C, Dimopoulos MA, D´Sa S, et al. Consensus treatment recommendations from the tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematol. 2020;7(11):e827-e837. doi:10.1016/S2352-3026(20)30224-6.
10. Kastritis E, Kyrtsonis MC, Morel P, Gavriatopoulou M, Hatjiharissi E, Symeonidis AS, et al. Competing risk survival analysis in patients with symptomatic Waldenström macroglobulinemia: the impact of disease unrelated mortality and of rituximab-based primary therapy. Haematologica. 2015;100(11):e446-9. doi:10.3324/haematol.2015.124149.
11. Kristinsson SY, Koshiol J, Björkholm M, Goldin LR, McMaster ML, Turesson I, et al. Immune-related and inflammatory conditions and risk of lymphoplasmacytic lymphoma or Waldenstrom macroglobulinemia. J Natl Cancer Inst. 2010;102(8):557-67. doi:10.1093/jnci/djq043.
12. McShane CM, Murray LJ, Engels EA, Anderson LA. Community-acquired infections associated with increased risk of lymphoplasmacytic lymphoma/Waldenström macroglobulinaemia. Br J Haematol. 2014;164(5):653-8. doi: 10.1111/bjh.12671.
13. Lund SH, Hultcrantz M, Goldin L, Landgren O, Björkholm M, Turesson I, et al. Patterns of Infectious Morbidity in Patients with Waldenströms Macroglobulinaemia / Lymphoplasmacytic Lymphoma: A Population-Based Study. Blood 2014;124(21):3350. doi:https://doi.org/10.1182/blood.V124.21.3350.3350