Introdução:
A hemofilia A adquirida é uma doença rara que se caracteriza pela presença de inibidores adquiridos contra o fator VIII da coagulação, ausentes no nascimento. Afeta igualmente homens e mulheres. A incidência é de 1 a 4 por milhão/ano, distribuída por dois picos etários, entre os 20 e os 30 anos, e entre os 60 e os 80 anos.1
Os inibidores da coagulação são auto-anticorpos, geralmente imunoglobulinas policlonais da classe IgG, que neutralizam a ação do fator VIII. Associam-se frequentemente a doenças autoimunes, como o Lúpus Eritematoso Sistémico e Artrite Reumatóide.1,2
Em 10% dos doentes há evidência de doença hematológica maligna ou neoplasia sólida (particularmente do pulmão e da próstata). A patofisiologia subjacente a estes casos não está completamente definida, podendo relacionar-se com fenómenos imunológicos induzidos por antigénios derivados do tumor.1,2
Clinicamente, manifesta-se com hemorragias espontâneas em doentes sem história pessoal ou familiar de coagulopatia. A maioria dos doentes apresentam-secom hematomas subcutâneos espontâneos e hematomas extensos das mucosas e tecido celular subcutâneo, e, menos frequentemente, com hemartroses, contrariamente ao que acontece no défice congénito.3
O diagnóstico laboratorial baseia-se na presença de prolongamento isolado do tempo parcial de tromboplastina ativada (aPTT), associado à redução do FVIII com presença de atividade do inibidor de FVIII. O tempo de protrombina e a função plaquetária estão normais.1,3
Caso Clínico:
Descreve-se um caso de uma mulher de 90 anos, com queixas com um mês de evolução de palidez cutânea e astenia de agravamento progressivo. Era referida história de equimoses e hematomas espontâneos, sem traumatismo associado, e um episódio de dejeção compatível com melenas. Apresentava antecedentes pessoais de hipertensão arterial medicada e controlada.
No exame objetivo, aquando da admissão, era evidente palidez da pele e mucosas, hematoma mandibular esquerdo, hematoma no joelho direito com edema na região rotuliana. Naauscultação cardíacaera audível um sopro sistólico grau III/VI.
Dos exames complementares realizados, apresentava anemia grave, com hemoglobina 4,7g/dL, hipocrómica, com ferropenia (Ferro 25mg/dL, capacidade de fixação de ferro de 290, ferritina 76mg/dL e saturação de transferrina de 8,6%), prolongamento do aPTT (67,1 segundos para um normal do dia de 28,2 segundos) com tempo de protrombina normal, lactato desidrogenase 296mg/dL e Velocidade de Sedimentação de 40mm/1ª hora. Realizou radiografia do joelho direito, sem evidência de sinais de fratura óssea, seguida de artrocentese que excluiu hemartrose, com extração de pequeno volume de líquido sinovial não hemático.
Foi considerada hipótese inicial de hemofilia adquirida, e confirmada após doseamento de atividade de FVIII de 2% e título elevado de inibidor -12 Unidades de Bethesda (UB) (uma UB corresponde à quantidade de anticorpos circulantes capazes de inativar 50% do fator VIII no plasma normal após incubação4). Apesar da necessidade de suporte transfusional e de apresentar hematomas espontâneos durante o internamento, nomeadamente do bicípede braquial (Fig. 1) e equimoses extensas do membro superior direito e da parede tóraco-dorsal homolateral (Fig. 2), a doente manteve estabilidade clínica, sem hemorragias ameaçadoras de vida. Iniciou corticoterapia 1mg/Kg/dia com boa resposta e normalização do aPTT, desaparecimento do inibidor e normalização da atividade de FVIII após um mês de tratamento.
Do estudo etiológico, não havia descrição de história familiar de hemofilia. Foram excluídas causas farmacológicas (antibióticos como penicilinas e sulfonamidas, anticonvulsivantes como fenitoína, e outros fármacos como a metildopa) e infeciosas (VIH, hepatite B e C). Apesar de ausência de clínica sugestiva de doença auto-imune, realizou estudo imunológico, com fator reumatóide e anticorpos antifosfolipídicos negativos, anticorpos antinucleares com título <1:160 com ponteado nuclear e citoplasmático, anticorpo anti-dsDNA e ENA negativos, sem consumo de complemento. Para exclusão de doenças neoplásicas sólidas ou hematológicas, foram realizados os seguintes exames complementares de diagnóstico, doseamento de imunoglobulinas, eletroforese de proteínas séricas, imunoeletroforese de proteínas séricas e tomografia computorizada tóraco-abdomino-pélvica que não mostraram alterações. Realizou ecografia tiroideia, que denunciou um nódulo hiperecogénico com halo hipoecogénico, de 25x16milimetros. Não foi realizado estudo dirigido ao nódulo tiroideu, nem estudos endoscópicos, por recusa da doente, mas não se objetivaram perdas hemáticas gastrointestinais e a função tiroideia estava normal. A doente acabou por falecer por intercorrência infeciosa, um mês após início de terapêutica.
Discussão:
Pela sua raridade, o diagnóstico de hemofilia adquirida é um desafio clínico. Pode levar a hemorragias ameaçadoras de vida, com hematomas retroperitoneais e síndromes compartimentais, o que requer um reconhecimento e diagnóstico atempados. A taxa de mortalidade global atinge os 33%.4-6
Neste caso, a presunção da hemofilia A ser adquirida baseou-se na ausência de história pessoal prévia de hemorragias e de história familiar.1,6
Em 50% dos casos, particularmente nos idosos, o desenvolvimento de auto-anticorpos contra o fator VIII é idiopático. Nos restantes 50%, encontram-se as doenças autoimunes, a gravidez, fármacos e malignidade, sendo esta última responsável por 10%.5
Apesar de não ter sido estabelecida a etiologia para a hemofilia adquirida no caso apresentado, os autores consideram a neoplasia como a mais provável neste caso, nomeadamente do tubo digestivo, explicando simultaneamente a anemia ferropénica. É discutível se teria tido interesse para a doente realizar os estudos endoscópicos, que provavelmente não teriam alterado as atitudes terapêuticas.
O tratamento desta entidade tem como objetivos a hemostase e a erradicação dos inibidores da coagulação.5-8O tratamento de primeira linha para a erradicação do inibidor é a corticoterapia isolada ou em associação a ciclofosfamida ou rituximab, consoante o título de inibidores e atividade do FVIII. Esta associação terapêutica deve ser ponderada de acordo com as características do doente, uma vez que a sua toxicidade também é superior.
Nos doentes com alto título de inibidores e hemorragia grave, podem ser usados agentes hemostáticos, como o complexo protrombínico ativado, o fator VII recombinante ativado, ou mesmo recorrer a técnicas de remoção como a plasmaferese.Quando o nível de inibidores se mantém elevado ou os níveis de fator VIII baixos após 4-6 semanas de tratamento, devem ser equacionadas terapêuticas de segunda linha, nomeadamente o anticorpo monoclonal anti CD 20 ou a ciclofosfamida.5,6,7,8Quando os títulos dos inibidores são baixos (<5UB), a desmopressina e o fator VIII recombinante são opções a considerar.
No caso apresentado, apesar da boa resposta à corticoterapia a doente acabou por falecer por causas infeciosas, responsáveis por 50% da mortalidade associada a esta patologia.5
A taxa de recorrência é de 20-30%. No seguimento, o aPTT e fator VIII devem ser monitorizados mensalmente nos primeiros 6 meses.6 Os estudos têm demostrado uma pior resposta à terapêutica com níveis elevados de inibidores e baixos níveis de fator VIII.5,7
Figura I
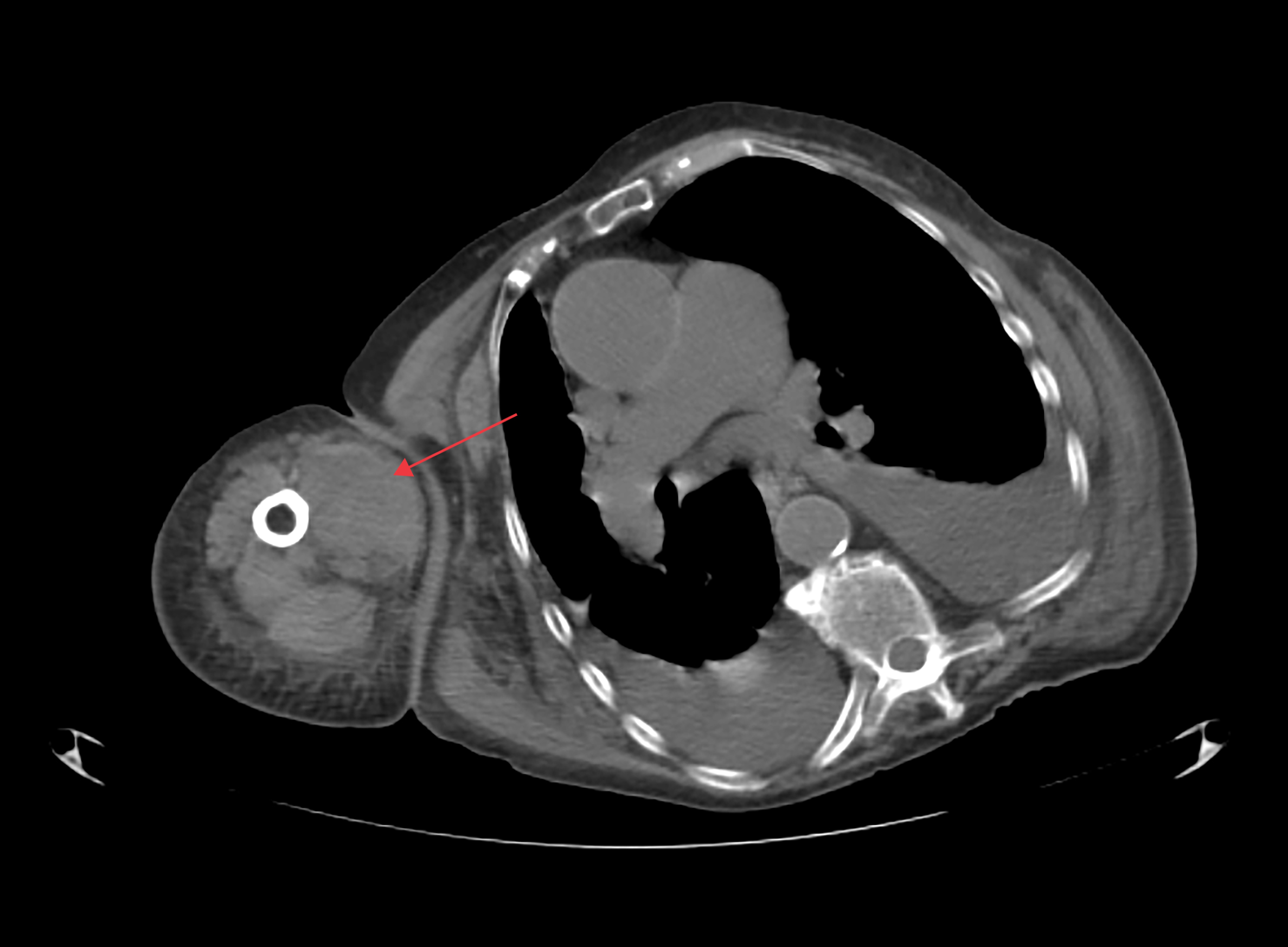
Tomografia computorizada a mostrar hematoma do bicípede braquial (seta).
Figura II
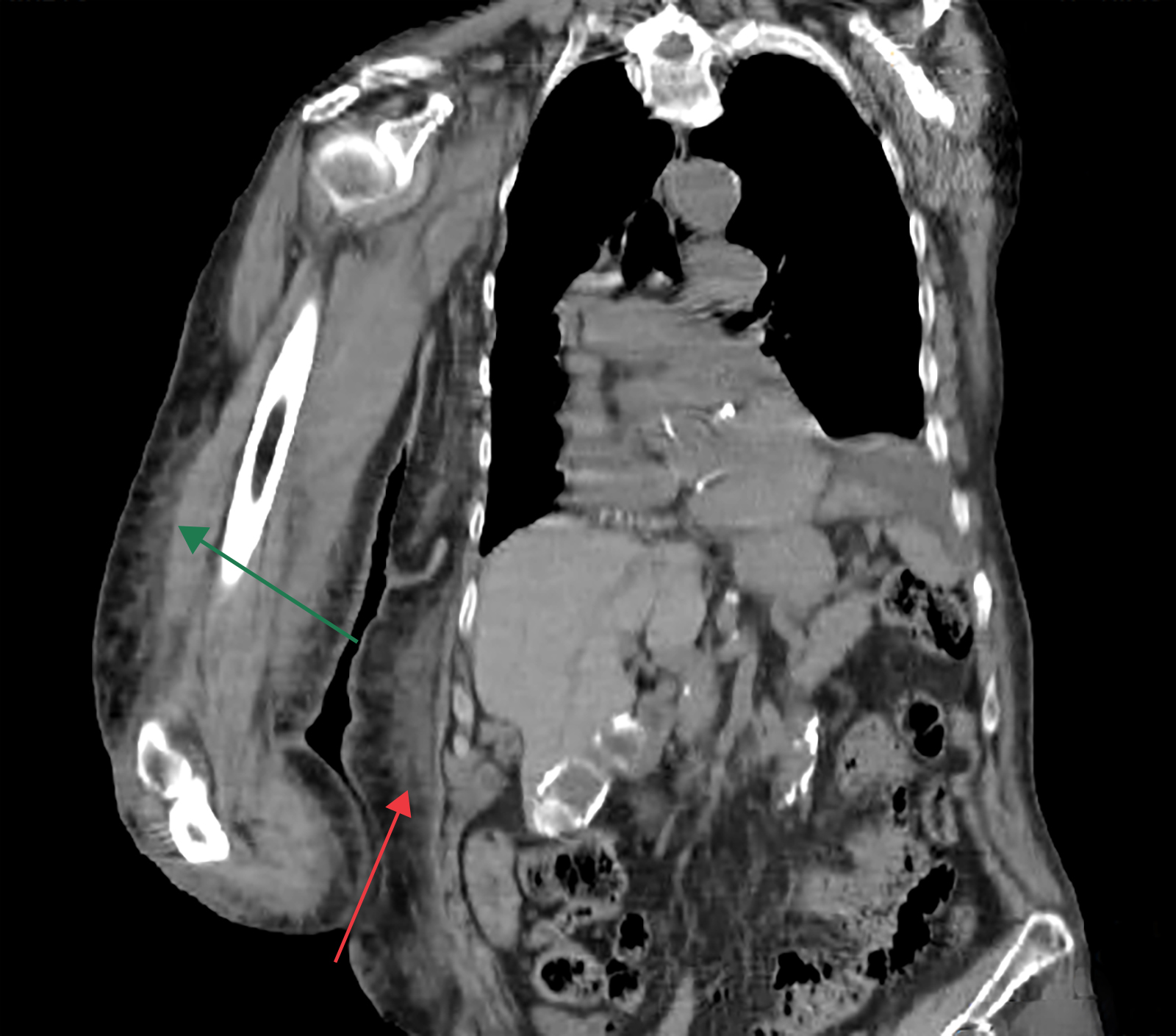
Tomografia computorizada a mostrar densificação da gordura do tecido celular subcutâneo do membro superior direito (seta verde) e parede tóraco-dorsal homolateral (seta vermelha).
BIBLIOGRAFIA
1 Elezović I. Acquired haemophilia syndrome: pathophysiology and therapy. Srp Arh Celok Lek. 2010 Jan;138 Suppl 1:64-8.
2 Buczma A, Windyga J. Acquired haemophilia. Pol Arch Med Wewn. 2007 May-Jun;117(5-6):241-5.
3 Janbain M, Leissinger CA, Kruse-Jarres R. Acquired hemophilia A: emerging treatment options. J Blood Med. 2015;6:14350.
4 Lan Mo and George C. Bao. Acquired factor VIII deficiency: two case reports and a review of literature. Exp Hematol Oncol. 2017; 6: 8.
5 Sakurai Y, Takeda T. Acquired hemophilia A: a frequently overlooked autoimmune hemorrhagic disorder. J Immunol Res. 2014;2014:320674.
6 García-Chávez J, Majluf-Cruz A. Acquired hemophilia. Gac Med Mex. 2020;156(1):67-77. English.
7 Charlebois J, Rivard GÉ, St-Louis J. Management of acquired hemophilia A: Review of current evidence. Transfus Apher Sci. 2018 Dec;57(6):717-20.
8 Tiede A, Collins P, Knoebl P, Teitel P, Kessler C, Shima M, et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica 2020;105(7):1791-801.

