Introduction:
Thrombotic thrombocytopenic purpura (TTP) is a rare and potential fatal thrombotic microangiopathy, that results from either a congenital or acquired decrease or absence of the enzyme a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13 (ADAMTS13).1 In this setting theres a predisposition to platelet aggregation which causes disseminated microthrombi in microcirculation leading to end organ damage, the most common being central nervous system and kidneys.1 The classic pentad consists in thrombocytopenia, microangiopathic hemolytic anemia (MAHA), fever, neurologic abnormalities and renal failure. Several cohort studies have demonstrated that less that 10% of patients diagnosed with acute TTP experience all five clinical features.2 Peripheral blood smear (PBS) usually presents schistocytes (erythrocyte fragmentation) formed after destruction of red cells in small vessels that are partially occluded by thrombi.1
Due to the fact that clinical presentation on TTP can be heterogeneous, a high index of suspicion is required for an early diagnosis, with laboratory findings playing an essential role. First line therapy of acquired TTP consists of plasma exchange (PEX), and it should be promptly initiated. The outcomes of patients depend on age, presence of neurological deficits, renal dysfunction, response to treatment and the presence of comorbidities.3
Case Description:
A 34-year-old caucasian female, with no significant medical or surgical history (including no prior SARS-CoV2 infection or immunization), nor daily medication, was admitted to the emergency department with complaints of fatigue for mild efforts and spontaneous hematomas with one week evolution. She also reported a self-limited episode of diarrhea and difficult-to-treat headache 15 days before. Besides scattered non-traumatic hematomas, she noticed a heavier menstrual flow than usual denying other sources of blood loss, namely epistaxis, gingival bleeding, hematuria or gastrointestinal losses. Gastrointestinal and urinary symptoms, fever or other neurological complaints were denied by the patient. On physical examination, she presented pale skin, small bruises in the upper limbs, with no purpura or petechiae; vital signs were within normal range (blood pressure 127/70mmHg, heart rate 77/min, afebrile). Neurological, cardiovascular, and respiratory examination were unremarkable. Full blood count revealed hemoglobin of 6.7g/dL (normal range (NR) 11.5-16.5 g/dL), MCV of 98.3fL (NR 76.0 - 96.0fL), with marked reticulocytosis (16.80%, NR 0.50-2.00%), platelets count of 8x109/L (NR 150-500x109/L) and normal white blood cell count. Peripheral blood smear showed marked thrombocytopenia and policromasia with few schistocytes (Fig. 1). Further investigations revealed normal prothrombin time, activated partial thromboplastins time and fibrinogen, INR of 1.0 (NR 0.8-1.1) D-dimer 2675ng/mL (NR <500ng/mL), lactate dehydrogenase of 909U/L (NR 120-246U/L), total bilirubin 2.05mg/dL (NR 0.30-1.20mg/dL) with indirect bilirubin of 1.46mg/dL (NR 0.10-1.00mg/dL), renal function with creatinine of 0.90mg/dL (NR 0.50-1.10mg/dL) and blood urea slightly increased (55.7mg/dL, NR 19-51mg/dL). Haptoglobin was decreased, direct antiglobulin test (DAT) and urinary hemosiderine were negative. The patient underwent a transfusion of one unit of fresh frozen plasma (FFP) and one unit of packed red blood cells (with partial hemoglobin recovery (9.0g/dL)), with maintained clinical stability. Considering clinical presentation (fatigue, hematomas, and menstrual losses) and lab results (DAT-negative hemolytic anemia, thrombocytopenia and schistocytes in PBS) the diagnosis of TTP was hypothesized. We then applied Plasmic score for TTP which was consistent with an intermediate risk (score 5) of having TTP and performed ADAMTS13 assay study in order to confirm our suspicion. The patient immediately initiated PEX plus prednisolone 1mg/kg/day. Meanwhile, HIV/HCV/HBV serologies, PCR SARS-CoV2 test and autoimmunity study were negative, abdominal ultrasound and head computed tomography found no relevant alterations and we were informed that pre-exchange ADAMTS13 activity was 0% (NR 40-140%) and ADAMTS 13 antibody of 95U/mL (NR <15U/mL), confirming the diagnosis of idiopathic acquired TTP.
After 13 sessions of PEX associated with corticotherapy, hemoglobin and platelet count progressively returned to normal (Fig. 2). At the time of discharge, the patient had complete recovery of symptoms and normalization of platelets count and hemoglobin level (hemoglobin 12.1g/dL, platelets 189 x109/L, ADAMTS13 activity was 103%). Prednisolone was gradually tapered down. After three months, the patient kept a favorable evolution without recurrence of symptoms, hemolytic anemia, and thrombocytopenia.
Discussion:
TTP remains a life-threatening condition with 5-16% mortality rate notwithstanding of appropriate and timely first-line treatment.3 Acquired TTP (aTTP) is the most prevalent form of the disease (~95%) and it is mainly due to the presence of anti-ADAMTS13 autoantibodies, typically presenting in adulthood.3,4 Genetic predisposing factors have been recognized in the development of aTTP such as female gender, obesity and black ethnicity,1,5 being the first one the only recognized in this case report.
Around 50% of TTP cases are triggered by severe infection, autoimmune diseases, HIV or, rarely, with drugs, cancer or after organ transplant (secondary TTP, sTTP). However, in about 49% of the situations, it is not possible to stablish a causal relationship (idiopathic TTP, iTTP), as in the reported case.2,6
Since January 2020, the SARS-CoV-2 pandemic has warned us to the several manifestations of COVID-19 infection, namely thrombotic complications. In fact, some studies have described the development of either SARS-CoV-2 infection or vaccination related TTP. SARS-CoV-2 infection-associated TTP seems to be related to direct damage of the endothelium and to the cytokine storm related to the infection.7 Vaccine-associated TTP is poorly understood, with a possible association with rise of anti-ADAMTS13 autoantibodies induced by the vaccine, with still few cases reported in the literature.8 In the presented case, both factors were declined, since clinical and analytical findings (mainly PCR test to SARS-CoV-2) were not suggestive of COVID-19 infection and the patient was not vaccinated against this disease.
Our patient presented symptoms related with anemia and thrombocytopenia, such as fatigue for mild efforts, spontaneous hematomas and higher menstrual losses, and headache which was interpreted as a neurological finding. Concomitantly, she reported one episode of diarrhea which can or not be related to the disease, since other gastrointestinal symptoms werent present (eg. abdominal pain, vomiting or nausea). Of note, recent literature reports an association between iTTP and digestive symptoms.3,6 Mariotte et al., in a cross-sectional analysis including 772 patients with adult-onset TTP, revealed that patients presenting iTTP had more neurological symptoms, lower frequency of renal impairment and more severe thrombocytopenia.6Neurological symptoms can occur at presentation, ranging from headache, confusion to major neurological abnormalities (stroke or coma).1,3 In fact, studies reported an incidence of neurological manifestations ranging from 60-80% in patients with TTP, being headache or confusion presented in nearly 30%.5,6
The timing of diagnosis and treatment of TTP determines the disease prognosis and mortality. The Plasmic score is a pretest probability model that predicts ADAMTS-13 deficiency in suspected TTP with high discrimination. It includes the evaluation of 7 variables, counting 1 point each: platelet count < 30 x 109/L, presence of hemolysis, no active cancer, no history of solid-organ or stem-cell transplant, mean corpuscular volume <90fL, INR<1.5 and creatinine <2.0mg/dL. A patient with intermediate or high risk (≥5 points) correlates with severe ADAMTS-13 deficiency and predicts a high rate of response to PEX.9 The fact that the patient presented a DAT-negative hemolytic anemia, thrombocytopenia and schistocytes in PBS along with severe ADAMTS13 deficiency (with <10% activity and 95% of anti-ADAMTS13 autoantibodies), confirmed the diagnosis of aTTP.
The cornerstone of the front-line therapy of aTTP is PEX with FFP replacement which must promptly be initiated. The exact duration of therapy is not predetermined and PEX should be daily performed until clinical and analytical response are achieved and sustained for 2 days.10Immunosuppressive therapy is also fundamental as adjunctive treatment, and it is normally started concomitantly with PEX.The addition of corticosteroids to PEX is recommended for patients with immune-mediated TTP since it has shown a reduction in mortality. The decision to initiate rituximab should be individualized and considered specially in patients with known comorbid autoimmune disease or in those who are in remission but still have low ADAMTS13 activity. Rituximabs benefit in preventing TTP relapse should also be taken into account. Caplacizumab is a humanized immunoglobulin fragment approved in the treatment of aTTP in association with PEX and immunosuppressive therapy. Due to its high cost and accessibility issues, it is usually indicated in patients with multi-organ involvement indicating a more severe disease or refractory patients who do not respond well to previous treatment for a given time period, as determined by a specialist physician with expertise in treating aTTP.10,11
In our patient, PEX plus corticotherapy were initiated during the first 24h after aPTT diagnosis with a good platelet and hemoglobin response and normalization of ADAMTS13 activity.
Conclusion:
TTP is a life-threatening condition, potentially fatal if not treated in a timely manner. With diverse array of clinical manifestations, an elevated index of suspicion is essential for prompt diagnosis, especially in patients with mild and unspecific symptoms.
Figura I
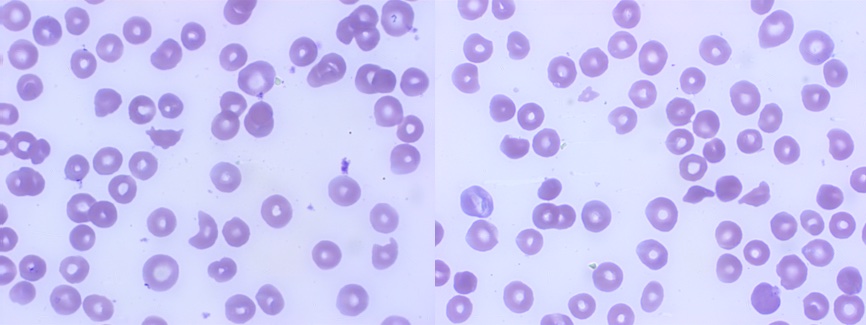
Peripheral blood smear showing schistocytes
Figura II
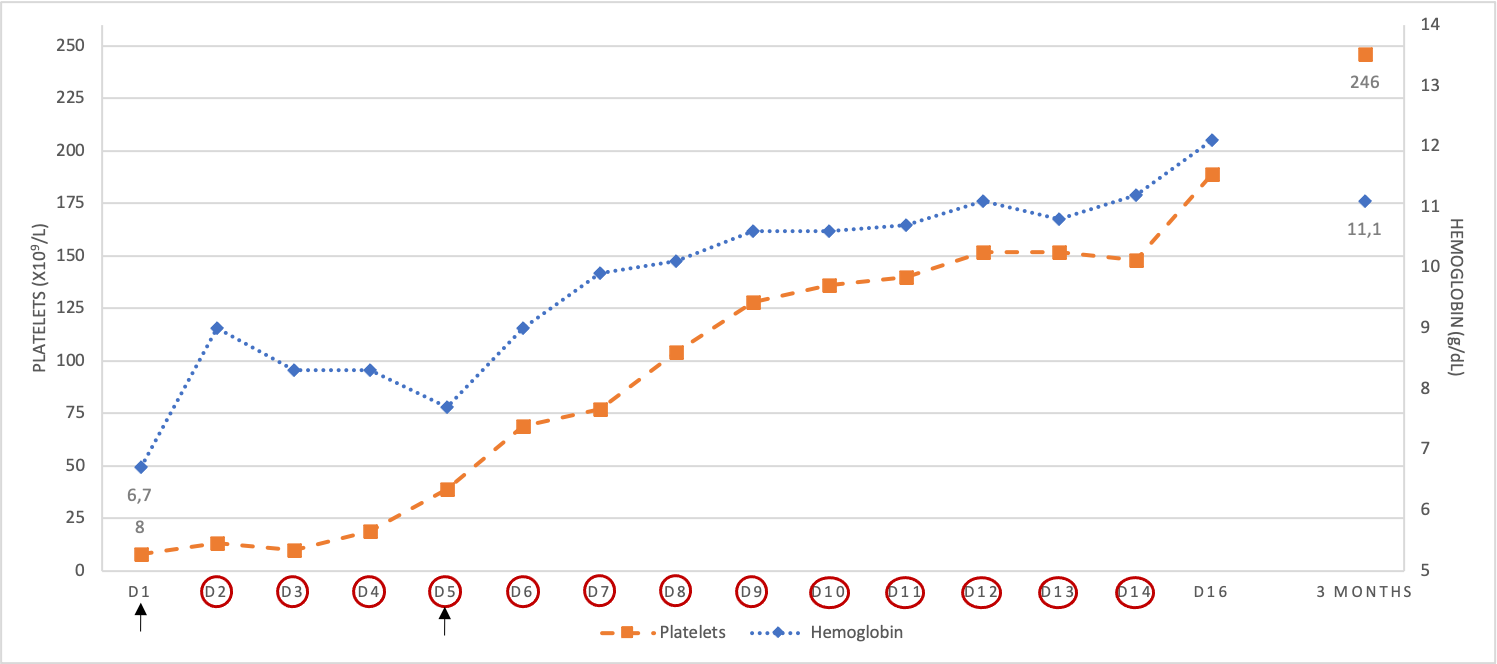
Graphic showing time evolution of platelet count and hemoglobin during the course of treatment. Circles indicate the day when treatment of PEX was performed. Arrows indicate the days that transfusion of blood components occurred.
BIBLIOGRAFIA
1. Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood 2017;130(10):1181-8. doi: 10.1182/blood-2017-04-636431
2. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017; 129(21): 283646. doi: 10.1182/blood-2016-10-709857.
3. Chiasakul T, Cuker A. Clinical and laboratory diagnosis of TTP: an integrated approach. Hematology Am Soc Hematol Educ Program 2018;1:530-8. doi: 10.1182/asheducation-2018.1.530.
4. Joly BS, Coppo P, Veyradier A. An update on pathogenesis and diagnosis of thrombotic thrombocytopenic purpura, Expert Review of Hematology 2019;12(6):383-95. doi: 10.1080/17474086.2019.1611423
5. Swart L, Schapkaitz E, Mahlangu JN. Thrombotic thrombocytopenic purpura: A 5-year tertiary care centre experience. J Clin Apher 2019;34(1):44-50. doi: 10.1002/jca.21673.
6. Mariotte E, Azoulay E, Galicier L, Rondeau E, Zouiti F, Boisseau P et al.A French Reference Center for Thrombotic Microangiopathies. Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): a cross-sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol 2016;3(5):e237-45. doi: 10.1016/S2352-3026(16)30018-7.
7. Tehrani HA, Darnahal M, Vaezi M, Haghighi S. COVID-19 associated thrombotic thrombocytopenic purpura (TTP); A case series and mini-review. Int Immunopharmacol. 2021;93:107397. doi: 10.1016/j.intimp.2021.107397
8. Alislambouli M, Victoria AV, Matta J, Yin F. Acquired thrombotic thrombocytopenic purpura following Pfizer COVID-19 vaccination. eJHaem. 2021;1-4.https://doi.org/10.1002/jha2.342
9. Bendapudi PK, Hurwitz S, Fry A, Marques MB, Waldo SW, Li A, Sun L, Upadhyay V, Hamdan A, Brunner AM, Gansner JM, Viswanathan S, Kaufman RM, Uhl L, Stowell CP, Dzik WH, Makar RS. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;4(4):e157-64. doi: 10.1016/S2352-3026(17)30026-1.
10. Sukumar S, Lämmle B, Cataland SR. Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management. J Clin Med 2021;10(3):536. doi: 10.3390/jcm10030536.
11. Zheng XL, Vesely SK, Cataland SR, Coppo P, Geldziler B, Iorio A. et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2496-502. doi: 10.1111/jth.15010.

